
Frontiers in Biomedical Sciences, Vol. 1, No. 2, November 2016 Publish Date: Sep. 23, 2016 Pages: 31-38
Role of P-glycoprotein in Chemotherapeutic Drug Resistance and Mechanisms of Pump Deactivation to Overcome MDR in Cancer Cells–A Critical Review
Proma Chakraborty1, 2, Mani Ramakrishnan1, 3, *
1Department of Biotechnology, Reva University, Rukmini Knowledge Park, Yelahanka, Bangalore, India
2Department of Life Sciences, Garden City College of Science and Management Studies, Bangalore, India
3PG Department of Bioscience, CMR Institute of Management Studies, OMBR Layout, Banaswadi, Bangalore, India
Abstract
P-glycoprotein (P-gp) is considered to be the multi-drug resistance integral membrane bound protein with a size of 170kDa, responsible for energy dependent efflux transport through ATP hydrolysis. In the normal and even in cancer cells, physiologically the pump prevents the destructive presence of poisons, drugs and xenobiotics to the body by expelling them out and maintaining the blood brain barrier. P-gp is found to play the fundamental regulator role in the pharmacokinetic properties of numerous clinically imperative therapeutic agents. The designing of cancer chemotherapy has turned out to be progressively modern, yet there is no malignancy treatment that is 100% successful against dispersed growth. Resistance to treatment with anticancer drugs results from a variety of factors which mainly includes polymorphic variations and genetic differences in tumours. The most well known explanation behind obtaining resistance to a broad range of anticancer drugs is influenced by the expression of one or more energy-dependent transporters (P-gp pumps or multi drug transporter) that detect and eject anticancer drugs from cells. A number of potential chemicals commonly known as inhibitors has been identified that can deactivate P-gp but has adverse effect on the host.
Keywords
P-glycoprotein, Multidrug Resistance (MDR), Drug Transporters, P-glycoprotein Inhibitors
Received: August 14, 2016
Accepted: August 31, 2016
Published online: September 23, 2016
@ 2016 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY license. http://creativecommons.org/licenses/by/4.0/
Contents
1. Introduction 2. Structure of P-glycoprotein 3. Steps Involved in Drug Efflux 3.1. Drug/Binding Recognition 3.2. ATP-Binding and Subsequent Hydrolysis 3.3. Efflux of Substrate/Drug Through Central Pore 4. Role of P-glycoprotein in Chemotherapeutic Resistance 5. Effects of P-glycoprotein Inhibition 5.1. Resistance Modifying Agents (RMAs) 5.2. Inhibition by the Cyclosporin Derivatives 5.3. Pharmacological Agents Which Act as P-gp Substrates Cum Inhibitors 5.4. Plant Derived Polyphenolic Compounds 5.5. Other Methods of Pump Deactivation 6. In-Silico Tools to Predict P-gp Inhibition 7. Conclusion
1. Introduction
The lipid bilayers of biological membrane are impermeable to ions and polar molecules. These molecules cannot cross the membrane by simple diffusion so their permeability is conferred by two classes of membrane proteins, pumps and channels. Pumps are commonly known as energy transducers; they use ATP to drive the thermodynamically uphill transport of ions or macromolecules. There are two types of ATP-driven pumps, P-type ATPases and the ATP-binding cassette pumps, they undergo conformational changes on ATP binding and hydrolysis that cause a bound ion or molecule to be transported across the membrane [1]. There are time to time reports on the P-gp structure and its role in drug molecule transport and its mechanisms (Table 1). P-gp is a plasma membrane protein which has a localized drug transport mechanism, actively exporting drugs out of the cell thus helping in distribution metabolism and excretion of drugs. These pumps are also involved in removal of toxic xenobiotics by extruding them out of the cell and excreting these compounds into urine and bile. Thus these transporters functions as a biological barrier preventing harmful accumulation of drugs and xenobiotic compounds in our body [2]. Drug transport requires ATP hydrolysis and actively generates a drug concentration gradient. The drug binding site of p-gp is conformationally coupled to the nucleotide-binding domains (NBDs) thus binding of drugs and ATP induces conformational changes in the protein, and it act as a drug flippase, moving drugs from the inner to the outer leaflet of the bilayer. P-gp also has a broad specificity for hydrophobic substrates, including many chemotherapeutic drugs [3].
Table 1. Research progresses on P-gp in chemotherapeutic drug resistance and mechanisms of pump deactivation to overcome such multidrug resistance in cancer cells.
2. Structure of P-glycoprotein
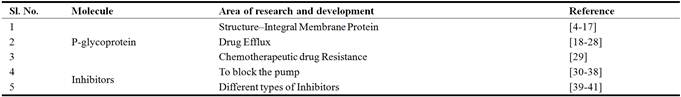
P-gp belongs to ATP binding cassette subfamily B (ABCB) of ATP–binding cassette (ABC) transporter [4, 5] and also commonly known as ABCB-1 or multidrug resistance–1 (MDR1) [8, 35]. It has two symmetrical halves or cassettes which are homologous in nature. Each of these cassettes contains six transmembrane domains followed by a nucleotide-binding domain (NBD). Homologous halves are separated by an intracellular flexible linker polypeptide loop that is approximately 75 amino acids in length [6, 7]. The connector region provides flexibility to the structure and helps in coordination between the two halves of the protein which is required for suitable contact of the two ATP binding sites [8]. P-gp has two ATP-binding domains which are known as nucleotide-binding folds (NBFs) present in the cytoplasm. NBFs transfer the energy to transport the substrates across the membranes and the ABC pumps are generally unidirectional.
The ATP-binding domains of P-gp have three regions, walker A, B, and signature C motifs. Highly conserved Lys residue inside the walker A motif (histadine permease) is directly associated with binding of ATP and an exceptionally rationed Asp residue inside the walker B motif serves to tie the Mg2+ion [9]. Human P-gp, the MDR1 gene product, requires both Mg2+-ATP-binding and hydrolysis to work as a drug transporter. It has likewise been recommended that magnesium may assume a part in stabilizing the ATP–binding site [10]. Signature C motif most likely participates to accelerate ATP hydrolysis by means of chemical transition state interaction [11] and also required in the transduction of the vitality of ATP hydrolysis to the conformational changes. This is limited to membrane integral domain necessary for translocation of the substrate [12].
ATP-binding sites are only restricted to walker A motifs of ATP binding domain with numerous substrate or drug binding sites in all through the transmembrane (TM) domain, intracellular loops and also ATP binding domain of P-gp. The major drug binding sites resides within TM6 and TM12 while other domains TM1, TM4, TM10, and TM11 also reported to have drug-binding sites [13, 14]. The TM1 amino acids are concerned in the development of a binding pocket that assumes a part in deciding the sensible substrate size for P-gp, whereas Gly build-ups in TMs2 and 3 are important in deciding substrate specificity. The nearby vicinity of transmembranes like TM2/TM11 and TM5/TM8 demonstrates that these locales between the two halves must encase the drug binding pocket at the cytoplasmic side of P-gp. They may shape the "pivots" required for conformational changes amid the transport cycle as well [15].
Each of the two TM areas of P-gp comprises of six long α-helical fragments. Five of the α-helices from every TM area are connected by a pseudo-twofold symmetry, though the sixth breaks the symmetry. The two α-helices situated nearest to the (pseudo) symmetry hub at the focal point of the particle have all the earmarks of being twisted [16]. P-gp has amino and carboxyl terminals. Initial theory suggested that N-terminal ATP-binding domain contains all residues required to hydrolyze ATP without interaction with the C-terminal ATP-binding domain [17]. Recent research confirms that both the amino-and carboxyl-terminal ATP binding sites can hydrolyze ATP [18] (Figure 1).
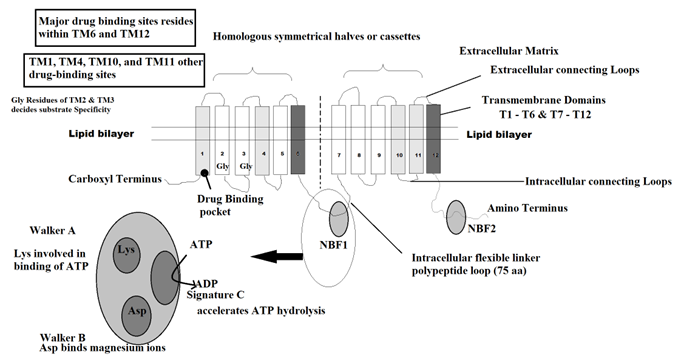
Fig. 1. Structure of p-glycoprotein pump.
3. Steps Involved in Drug Efflux
ABC transporters are expressed in the epithelium of the liver and intestine, where the proteins ensure the body by pumping drugs and other harmful substances into the bile duct and intestinal lumen. They additionally play an important role in maintaining the blood-brain barrier. Steps involved in drug efflux are as follows:
3.1. Drug/Binding Recognition
The significant drug binding sites are localized in or near TM6, TM12, TM1, TM4, TM10, and TM11. Amino acids in TM1 are involved in the development of a binding pocket that assumes a part in deciding the reasonable substrate/drug size for P-gp, whereas Gly residues in TMs 2 and 3 are critical in deciding substrate specificity. Consequently, these transmembrane areas help in substantial to perceive substrates/drugs.
3.2. ATP-Binding and Subsequent Hydrolysis
ATP–binding and its following hydrolysis are the fundamentals of drug transport utilizing 0.6–3 molecules of ATP for each drug molecule when transported outside the cell [19]. The recent reports suggest that two ATP molecules are utilised for the transport of every substrate particle [20]. One of the two ATP hydrolysis have role in solitary turnover of the catalytic cycle of P-gp in the transport of substrate and the other in affecting conformational changes to reset the pump for synergist cycle [21]. Both nucleotide-restricting domains act symmetrically and amid individual hydrolysis event, the ATP binding sites are selected in a random manner. In addition, only one nucleotide site hydrolyzes ATP at any given time, bringing about (in this site) a conformational change that radically diminishes (>30-fold) the fondness of the second site for ATP-binding. Subsequently, the blocking of ATP binding to the second site in spite of the fact that the first is in reactant adaptation is alluded as substitute reactant cycle of ATP hydrolysis and ADP release is the rate-constraining stride in the synergist cycle and the substrates apply their impact by regulating ADP release [22].
Moreover, according to Takada and Yamada (1998) and Urbatsch et al. (2003), the two nucleotide binding domain dimerize to outline an incorporated single body containing two bound ATP with one and only of the two ATP being hydrolyzed per dimerization event. In case one ATP-binding domain is not functional; there is no ATP hydrolysis even when ATP ties to other ATP-binding site [23, 24]. However, in spite of this, one study proposed that ATP hydrolysis by possibly one or both NBFs is crucial to drive transport of solutes. Transformations of either NBF1 or NBF2 decrease solute transport, yet don't cancel it totally. Thus, basic accord still must be made seeing ATP hydrolysis as one utilitarian unit [25].
3.3. Efflux of Substrate/Drug Through Central Pore
P-gp captures lipophilic drugs as they travel through the lipid bilayer and flips the molecule from inner leaflets to outer leaflet and finally to the extracellular matrix. The substrate/drug binds to C-terminal stimulates the hydrolysis of ATP into ADP + Pi that induce a P-gp conformational change resulting in a decrease of affinity for the substrate, which is transferred first to the N-terminal site and then released outside the cell [26, 27].
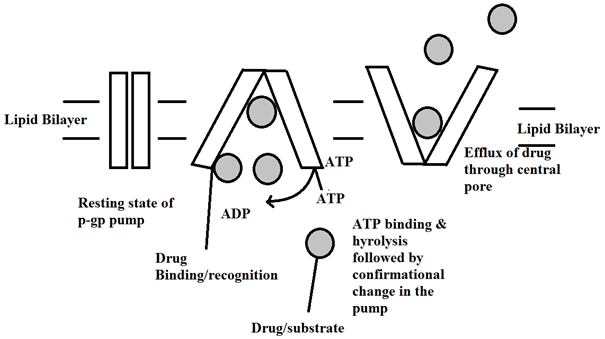
Fig. 2. Mechanism of drug efflux through the central pore of the p-gp pump.
When the nucleotide binds to the intracellular nucleotide–binding domain of p-gp the protein undergoes a conformational change. Signature C motif most likely partakes to accelerate ATP hydrolysis during which the membrane protein undergoes a conformational change in the integral membrane domain (Figure 3). This conformational change is required for the translocation of the substrate from inner to outer leaflet of lipid bilayer [12]. The transmembrane domains undergo a complete reorganization throughout the depth of the lipid bilayer on binding of nucleotide. On binding nucleotide, the TM domains reorder into three consolidated sections. Each section is 2-3 nm in diameter and 5-6 nm in depth. This restructuring opens a central pore along its length. This pore permits access of hydrophobic drugs from lipid bilayer to the central pore of the transporter. Recent research proposed that the drug substrates diffuse from the lipid bilayer into the drug binding pocket through "gates" first and ‘gates’ are framed by TM fragments at either end of drug binding pocket. They efflux the substrate through the central pore of the transporter outside the lipid bilayer and release them into the extracellular matrix [27-29].
4. Role of P-glycoprotein in Chemotherapeutic Resistance
Efflux via ABC transporters is a normal physiological process; involved in drug resistance in cancer cells. Three transporters—multidrug resistance protein 1 (MDR1), multidrug resistance associated protein 1 (MRP1), and breast cancer resistance protein (BCRP)—are mainly involved in many drug resistant cancers where they get over expressed in malignant cells [30]. All three transporters have broad substrate specificity and are able to efflux many chemotherapeutic drugs which includes vinca alkaloids, epipodophyllotoxins, anthracyclines, taxanes, and kinase inhibitors, from cells. Thus, induce resistance to the malignant cell line. MDR1, which produces P-gp, was the first of these to be distinguished and has been concentrated widely. Typical articulation of the MDR1 gene in the colon, liver, and kidney is expanded when these tissues get cancerous. Tissues that don't typically express MDR1, for example, lung, breast, and prostate cells, often become drug resistant due to expression of related transporters. Constitutive activation of signalling molecules like cyclins and cyclin dependant protein kinases drives the cell cycle out of control leading to uncontrolled proliferation of cells and thus results in cancer. Furthermore, these proteins likewise control P-gp expression and can in this manner regulate the environment to enable the development of drug resistance [31].
5. Effects of P-glycoprotein Inhibition
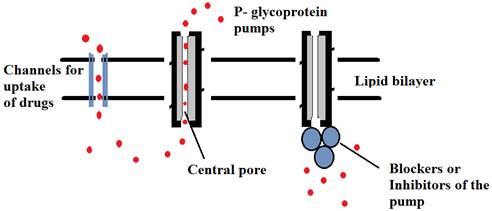
Fig. 3. Schematic representation of inhibitors blocking the substrate binding sites of P-glycoprotein pumps thus allowing the drug concentration in cell.
Chemotherapy would be more effective if P-gp pumps are shut off during treatment. Most endeavours to restrain P-gp have concentrated on the distinguishing proof of modulator exacerbates that repress P-gp action amid chemotherapy. Potential downsides of modulators are that they can induce/increase P-gp synthesis and maturation (folding) to the cell surface. Various techniques are used to concentrate on the structure and mechanism of how ATP hydrolysis is coupled to drug efflux. Studies including the folding of P-gp, determining the critical residues required in drug binding, deciding the number of drug binding sites, how substrates tie to P-gp and conformational changes that happen during drug binding and drug efflux give imperative insights that could be utilized to create methodologies to close down the pump amid chemotherapy. Various inhibitors are used to shut down the pumps [32] (Figure 3).
5.1. Resistance Modifying Agents (RMAs)
Advancement of biochemical modulators and utilization of the same with anti-cancer drugs is a present methodology of cutting edge disease chemotherapy. The greater parts of the reported resistance modifying agents (RMAs) are successful in vitro but have unfavourable impact on the hosts. Thus, the improvement of nontoxic RMA is of colossal significance in the field of cancer chemotherapy. A nontoxic resistance modifying agent oxalyl bis (N-phenyl) hydroxamic acid (OPHA) has been produced on the premise of the auxiliary shared traits of the reported RMAs. In a report, the restraint of P-gp by the compound, OPHA in human cervical malignancy cell line, Hela, has been portrayed by western blotting, study of immunofluorescence and enzyme linked immunofluorescence assay (ELISA). The high IC50 values (measure of the effectiveness of a substance in inhibiting a specific biological or biochemical function) of OPHA against various cell lines demonstrate the non lethal nature of the compound. It has likewise been tried in mice model yet its careful impact on human in vivo is yet to be affirmed [33].
5.2. Inhibition by the Cyclosporin Derivatives
The ATPase of P-gp is an imperative focus for MDR modulation, as the hydrolysis of ATP by P-gp is accepted to deliver the energy required for the dynamic transport of anticancer drugs. It is proposed that the inhibition by the cyclosporine derivatives of P-gp ATPase represents an alternative approach of action for these MDR modulators. Experiments with a selected series of anthracycline analogs of various positive charges, demonstrated that decidedly charged analogs are preferred perceived by P-glycoprotein over their neutral counterparts. The concentrate additionally demonstrated that analogs with increasing lipophilicity, regardless of charge, can hinder drug-binding to P-glycoprotein with more efficiency. These discoveries cleared up that electronic charge is an imperative variable for P-glycoprotein substrate transport and that lipophilicity is vital for MDR-reversing activity [17, 34]. One contrast in chemical structure between the cyclosporine derivatives and the stimulator used as a part of this study is that the stimulators are all cations while the cyclosporine derivatives are electronically neutral [35].
5.3. Pharmacological Agents Which Act as P-gp Substrates Cum Inhibitors
There are some pharmacological agents which act as P-gp substrates cum inhibitors. Verapamil, an antihypertensive calcium channel blocker, trifluoperazine, a calmodulin antagonist, cyclosporine, an immunosuppressant, different antihypertensives, for example, quinidine and reserpine, yohimbine, antiestrogenic tamoxifen and toremifene, and antineoplastic vincristine, all fall under the above classification. Since the greater parts of these compounds were P-gp substrates themselves, they collaborated with the protein, rivalled alternate substrates and went about as aggressive inhibitors. But all of these inhibitors have been non-particular and less intense. Their P-gp inhibitory fixations achieved high dangerous levels because of which a large portion of these inhibitors fizzled in clinical trials [36, 37]. The pharmacological agents which were P-gp substrates cum inhibitors were altered structurally viz. their chirality was adjusted to accomplish a superior or a null inherent pharmacological profile in order to reduce the toxicity of the parent compounds. Dexverapamil, the R-isomer of verapamil without any cardiac activity, PSC833 (valspodar), a cyclosporine A analogue which lacks the immunosuppressive character, MS-209 and several other drug derivatives or analogs fall under this class. These resultant modulators still remained P-gp substrates themselves and indicated low protein affinity but, their P-gp inhibitory measurement was a long ways past the tolerant dosage levels [38, 39]. Due to the chiral optimization, these chemosensitizers are dependent on cytochrome P4503A4 (CYP4503A4) substrates for metabolism, which made them simultaneously compete with the administered anticancer P-gp substrate drugs whose metabolism was also affected by the same system. This caused significant pharmacokinetic alterations that unpredictably affected the metabolic and clearance mechanisms of the substrate drugs thus making difficulties in adjusting the chemotherapy doses in patients. Every one of these issues left this class of inhibitors forsook [38, 39].
5.4. Plant Derived Polyphenolic Compounds
It was accounted for that plant derived polyphenolic compounds, predominantly flavonoids and stilbenes or their synthetic derivatives, can modulate the primary ABC transporters that are responsible for cancer drug resistance, including P-glycoprotein, multidrug resistance-related protein 1 (MRP1) and breast cancer resistance protein (BCRP). A few flavonoids and stilbenes have been accounted for to hinder BCRP encoded by the ABCG2 gene. In this manner, the utilization of flavonoids with high inhibitory activity could change pharmacokinetics and tolerance of drug levels that are effluxed by BCRP. Coumarins, terpenoids, alkaloids and saponins are some of the naturally occurring inhibitors which can be used successfully to modulate multi drug resistance in cancer cells [40, 41].
5.5. Other Methods of Pump Deactivation
The biological active conformation of a protein is highly required to retain its functional property. If the polypeptide chain of the desired protein is altered its functional property can be compromised. Chemical denaturant urea can be used successfully to denature alpha helical membrane proteins. When the denaturing agents are removed by dialysis, in presence of oxygen disulphide bonds and non-covalent bonds are reformed thus the protein gets back to its native state [42].
6. In-Silico Tools to Predict P-gp Inhibition
Label-powerset, binary relevance and classifiers chain are the three in-silico methods used to define the molecular features of the transporter. Label-powerset was used to build the model/pharmacological target depending upon the physiochemical property of the inhibitors. Then either classifiers chain or binary relevance is used to gain the predictability of the defined structure [43]. P-gp drug transporter is of great clinical significance. Different aspects of this protein including its functional domain, communicating ligands and its expression (tissue specific) need to be elucidated to enhance its therapeutic application [44].
7. Conclusion
There are various mechanisms to deactivate the drug efflux pumps of cancer cells using inhibitors that block the substrate binding sites and found that these inhibitors have toxic effects on other cells. Instead of deactivating the pump with an inhibitor, an attempt to alter the folding of p-glycoprotein might prevent the expression itself on the cell surface. The misfolded protein that does not reach the cell surface is retained in the endoplasmic reticulum (ER) and degrade rapidly. Thus, cancer cells can easily sustain the chemotherapeutic drugs followed by apoptosis or programmed cell death. Emphasis on deactivation of the expression of energy dependent drug efflux pump (P-glycoprotein pump) or the multidrug transporter by treating the cells with urea in the presence of the reducing agent β–mercaptoethanol would be an ideal technique through Anfinsen experiment. Urea is used to break non-covalent bonds i.e. hydrogen bonds. β–mercaptoethanol, a disulfide reducing agent can covalently interact with specific protein functional groups. Urea does not disrupt the membrane itself, but helps to solubilise the hydrophobic membrane protein in water. This chemical cocktail is ideal to use for unfolding the proteins. Native PAGE and Immunodetection techniques can be used for the studies like detection, identification and characterization of the unfolded (p-gp) protein. Furthermore, molecular dynamic simulation analysis revealed the pathway between multiple conformational states of the P-gp as well as molecular model at an atomic level that ascertain different parameters of the protein analysis.
References
- Berg J M, Tymoczko J L, Stryer L; Biochemistry; Chapter 13 Membrane Channels and Pumps; 5th edition; New York: W H Freeman; p 528-568 (2002).
- Linn J H, Yamazaki M; Role of P-glycoprotein in pharmacokinetics clinical implications. Clin Pharmacol 42: p 59–98 (2003).
- Sharom F J; The P-glycoprotein efflux pump: how does it transport drugs? J Membr Biol. 1; 160 (3): p 161-75 Dec (1997).
- Dean M, Rzhetsky A, Allikmets R; The human ATP-binding cassette (ABC) transporter superfamily. Genome Res; 11: p 1156–66 (2001).
- Venter J C, Adams M. D, Myers E W, Li P W, Mural R J, Sutton G G, et. al; The sequence of the human genome. Science; 291: p 1304–51 (2001).
- Gottesman M N, Pastan I; Biochemistry of multidrug resistance mediated by multi drug transporter. Annu Rev Biochem: 62: p 385–427 (1993).
- Loo T W, Clarke D M; Identification of the distance between the homologous halves of P-glycoprotein that triggers the high/low ATPase activity switch; J Biol Chem; 289 (12): p 8484-92 (2014).
- Hrycyna C A, Airan L E, Germann U A, Ambudkar S V, Pastan I, Gottesman M M; Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry; 37: p 13660–73 (1998).
- Hung L W, Wang I, Nikaido K; Crystal Structure of the ATP-binding subunits of an ABC transporter. Nature; 396: p 703–7 (1998).
- Booth C L, Pulaski L, Gottesman M M, Pastan I; Analysis of the properties of the N-terminal nucleotide-binding domain of human P-glycoprotein. Biochemistry; 39: p 5518–26 (2000).
- Tombline G, Bartholomew L, Gimi K, Tyndall G A, Senior A E; Synergy between conserved ABC signature Ser residues in P-glycoprotein catalysis. J Biol Chem; 279: p 5363–73 (2004).
- Yasuhisa K, Michinori M, Kei T, Tohru S, Noriyuki K; ATP hydrolysis dependent multidrug efflux transporter: MDR1/P-glycoprotein. Current Drug Metabolism; 5: p 1–10 (2004).
- Ambudkar S V, Dey S, Hrycyna; Biochemical, cellular and pharmacological aspects of multidrug transporter. Annu Rev Pharmacol Toxicol; 39: p 361–98 (1999).
- Ueda K, Taguchi Y, Morishima M; How does P-glycoprotein recognize its substrates. Semin Cancer Biol; 8: p 151–9 (1997).
- Tip W, Loo M, Claire B, David M C; Val 133 and Cys 137 in Transmembrane Segment 2 Are Close to Arg 935 and Gly 939 in Transmembrane Segment 11 of Human P-glycoprotein. J Biol Chem; 279: p 18232–38 (2005).
- Mark F R, Richard C, Szabolcs M, Christopher F H, Robert C F; Three dimensional Structure of P-glycoprotein The transmembrane regions adopt anasymmetric configuration in the nucleotide-bound state. J Biol Chem; 280: p 2857–62 (2005).
- Shimabuku A M, Nishimoto T, Ueda K, Komano T; P-glycoprotein. ATP hydrolysis by the N-terminal nucleotide-binding domain. J Biol Chem; 267: p 4308–11 (1992).
- Hrycyna C A, Ramachandra M, Ambudkar S V, Ko Y H, Pedersen P L, Pastan I, et.al;Mechanism of action of human P-glycoprotein ATPase activity. Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J Biol Chem; 273: p 16631–4 (1998).
- Ambudkar S V, Dey S, Hrycyna C A; Biochemical, cellular and pharmacological aspects of multidrug transporter. Annu Rev Pharmacol Taxicol; 39: p 361–98 (1999).
- Shapiro A B, Ling V; Stoichiometry of coupling of rhodamine 123 transporter to hydrolysis by human P-glycoprotein. Eur J Biochem; 254: p 189–93 (1998).
- Sauna Z E, Ambudkar S V; Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Nat l Acad Sci; 97: p 2515–20 (2000).
- Sauna Z E, Smith M M, Muller M, Kerr K M, Ambudkar S V; The mechanism of action of multidrug-resistance – linked P-glycoprotein. J Bioenerg Biomembr; 33: p 481–91 (2001).
- Takada Y, Yamada T Y; Non equivalent cooperation between the two nucleotide binding folds of P-glycoprotein. Biochim Biophys Acta; 1373: p 131–6 (1998).
- Urbatsch I L, Tyndall G A, Tombline G, Senior A E; P-glycoprotein catalytic mechanism studies of the ADP-vanadate inhibited state. J Biol Chem; 278: p 23171–9 (2003).
- Hou Y X, Cui L, Riordan J R, Chang X B; ATP binding to the first nucleotide binding domain of multidrug resistance protein MRP1 increases binding and hydrolysis of ATP and trapping of ADP at the second domain. J Biol Chem; 277: p 5110–9 (2002).
- Loo T W, Clarke D M; The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface; J Biol Chem; 274 (35): p 24759-65 (1999).
- Mark F R, Alhaji B K, Richard C, Christopher F H, Robert C F; Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J Biol Chem; 278: p 8294–99 (2003).
- Vishal R. Tandon, B. Kapoor, G. Bano, S. Gupta, Z. Gillani, S. Gupta, D. Kour; P-Glycoprotein: pharmacological relevance; Indian J Pharmacol; Vol 38; Issue 1; p 13-24: February (2006).
- Loo T W, Clarke D M; Do drug substrates enter the common drug-binding pocket of P-glycoprotein through "gates"? Biochem Biophys Res Commun; 329: p 419–22 (2005).
- Md. Lutful Amin; P-glycoprotein Inhibition for Optimal Drug Delivery; Drug Target Insights: 7; p 27–34 (2013).
- Genevieve Housman, Shannon Byler, Sarah Heerboth, Karolina Lapinska, Mckenna Longacre, Nicole Snyder and Sibaji Sarkar. Drug Resistance in Cancer: An Overview,Cancers,6, p 1769-1792; doi: 10.3390/cancers6031769; (2014).
- Loo T W, Clarke D M; The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface; J Biol Chem; 274 (35): p 24759-65 (1999).
- Choudhuri S K; Deactivation of P-glycoprotein by a novel compound, oxalyl-bis (N-phenyl) hydroxamic acid. Neoplasma, p 272-7, (2002).
- Lampidis T J, Kolonias D, Podona T, Israel M, Safa A R, Lothstein L, Savaraj N, Tapiero H & Priebe W; Circumvention of P-GP MDR as a function of anthracycline lipophilicity and charge. Biochemistry, 36, p 2679-2685 (1997).
- Toru Watanabe, Noriko Kokubu, Steven B, Charnick, Mikihiko Naito, Takashi Tsuruo & Dalia Cohen; Interaction of cyclosporine derivatives with the ATPase activity of human P-glycoprotein; British Journal of Pharmacology, 122, p 241–248 (1997).
- Kale Mohana, Raghava Srivalli, P. K. Lakshmi; Overview of P-glycoprotein inhibitors: a rational outlook; Brazilian Journal of Pharmaceutical Sciences vol. 48, n. 3, p 353-367, July/Sep (2012).
- Dantzig A H, De Alwis, D P, Burgess M; Considerations in the design and development of transport inhibitors as adjuncts to drug therapy. Adv. Drug Deliv. Rev, v. 55, p. 133-150 (2003).
- Darby R A, Callaghan R; McMahon R M; P-Glycoprotein inhibition; the past, the present and the future. Curr. Drug Metab, v. 12, p 722-731 (2011).
- Thomas H, Coley H M; Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control, v. 10, p 159-165 (2003).
- Pick A, Muller H, Mayer R, Haenisch B, Pajeva I K, Weigt M, et. al; Structure–activity relationships of flavonoids as inhibitors of breast cancer resistance protein (BCRP), Bioorganic & Medicinal Chemistry, Volume 19, Issue 6, p 2090–2102, 15 March (2011).
- Hossam M. Abdallah, Ahmed M. Al-Abd, Riham Salah El-Dine, Ali M. El-Halawany; P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review; Journal of Advanced Research, Volume 6, Issue 1, p 45–62, January (2015).
- Jana Broecker, Sebastian Fiedler, Sandro Keller; Complete and Reversible Chemical Unfolding of an α-Helical Membrane Protein; Biophysical Journal, Volume 104, Issue 2, Supplement 1, p62a, 29 January (2013).
- Floriane Montanari, Barbara Zdrazil, Daniela Diglesand Gerhard F. Ecker; Selectivity profiling of BCRP versus P ‑ gp inhibition: from automated collection of polypharmacology data to multi ‑ label learning; Journal of Cheminformatics 8: 7; February (2016).
- Veda Prachayasittikul, Virapong Prachayasittikul; P-Glycoprotein transporter in drug development; EXCLI Journal; 15–ISSN 1611-2156: p 113-118; February (2016).



