
Frontiers in Biomedical Sciences, Vol. 1, No. 1, September 2016 Publish Date: Aug. 19, 2016 Pages: 26-30
Conformational Analysis and Excited State Properties of a Competitive Inhibitor of Deoxyguanosine Triphosphate, 2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one (Ganciclovir)
F. J. Amaku, I. E. Otuokere*
Department of Chemistry, Michael Okpara University of Agriculture, Umudike, Nigeria
Abstract
Ganciclovir, (2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one) is an acyclic analog of the nucleoside guanosine. Ganciclovir is widely used for the treatment of cytomegalovirus (CMV) infections among patients with impaired cell-mediated immunity, particularly persons with poorly controlled and advanced HIV/AIDS and recipients of solid organ and bone marrow transplantation, who are at high risk for invasive CMV disease. Computer aided geometry optimization (active conformation) and excited state properties of Ganciclovir was performed by ArgusLab 4.0.1 software. Result showed that the best conformation of Ganciclovir was found to be -126.872353 au(-79613.675800 kcal/mol) which is the minimum potential energy calculated by ArgusLab software using AMI/RHF method. At this point, Ganciclovir will be more active to interact with the receptors. Such types of interactions are significant for drug- receptor interactions.
Keywords
Ganciclovir, ArgusLab, Geometry Optimization, Receptor
Received:May 20, 2016
Accepted: June 2, 2016
Published online: August 19, 2016
@ 2016 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY license. http://creativecommons.org/licenses/by/4.0/
1. Introduction
Ganciclovir is widely used for the treatment of cytomegalovirus (CMV) infections among patients with impaired cell-mediated immunity, particularly persons with poorly controlled and advanced HIV/AIDS and recipients of solid organ and bone marrow transplantation, who are at high risk for invasive CMV disease. Ganciclovir, (2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one) is an acyclic analog of the nucleoside guanosine [1]. The drug is converted intracellularly to ganciclovir 5'-monophosphate by a viral kinase, which is encoded by the cytomegalovirus (CMV) gene UL97 during infection [2]. Subsequently, cellular kinases catalyze the formation of ganciclovir diphosphate and ganciclovir triphosphate, which is present in 10-fold greater concentrations in CMV or herpes simplex virus (HSV)-infected cells than uninfected cells [3-4]. Ganciclovir triphosphate is a competitive inhibitor of deoxyguanosine triphosphate incorporation into DNA and preferentially inhibits viral DNA polymerases more than cellular DNA polymerases. In addition, ganciclovir triphosphate serves as a poor substrate for chain elongation, thereby disrupting viral DNA synthesis by a second route [5-6].
The geometry of a molecule has a great impact on its energy level, physical and chemical properties. As the molecule rotates, it adopts different conformations and spatial arrangements to achieve one of the stable states of lowest energy [4]. The total molecular energy can be evaluated in terms of potential energy surface as a sum of energies associated with each type of bonded interactions i.e. bond length, bond angle and dihedral angle as well as non-bonded interactions (van der Waals and electrostatic) taking place in a molecule and on atomic properties of a molecule [7, 8]. Computational study on the geometry optimization and excited – state properties using ArgusLab 4.0.1 software have been reported by several authors [20 – 24]. These authors reported the minimum energy of the drugs as the most feasible position the drug to interact with receptors. The present work describes the computer aided geometry optimization (active conformation) and calculation of excited state properties of 2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one,ganciclovir by ArgusLab 4.0.1 software [9].
2. Materials and Methods
2-Amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one (ganciclovir) structure was sketched with ACDLabChem Sketch software and saved as MDL molfiles (*mol). Ganciclovir structure was generated by ArgusLab [9], and minimization was performed with UFF (The Universal Force Field) molecular mechanics method [7-8]. The Universal Force Field is a molecular mechanics method. The method was first introduced in 1993 by Rappe and co-workers as a way to treat the entire periodic table. UFF is good for initially cleaning up structures that you have sketched in the builder, and for refining initial geometries before using more expensive methods [18]. The minimum potential energy was calculated using geometry convergence function in Argus lab software [9]. Surfaces created to visualize ground state properties as well as excited state properties such as orbital, electron densities, electrostatic potentials (ESP) spin densities and generated the grid data were used to make molecular orbital surfaces and electrostatic potential mapped on electron density surface [10-14]. The minimum potential energy was calculated for ganciclovir through the geometry convergence map [15]. Mulliken atomic charges and ZDO atomic charges of ganciclovir were determined using AM1 method [7,12].
3. Results
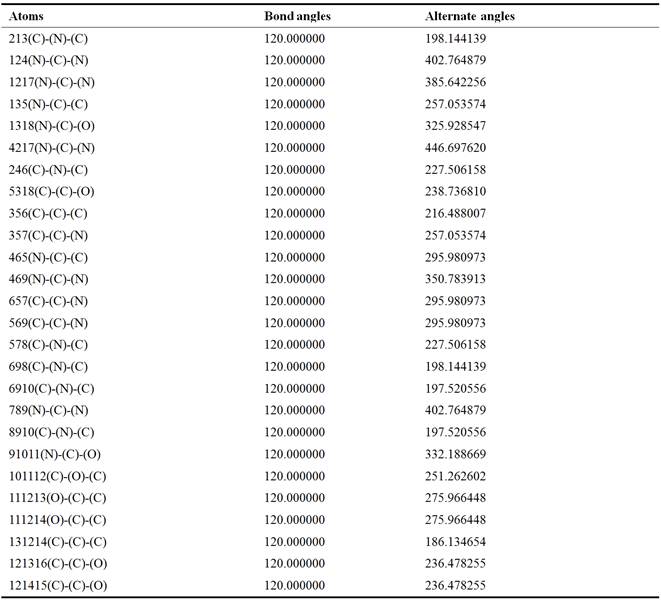
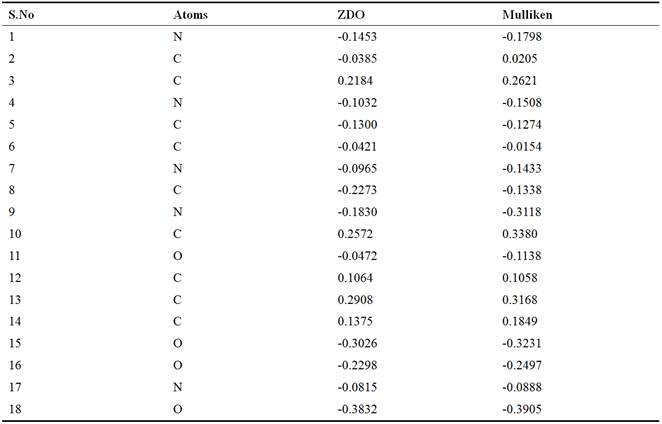
Prospective view and calculated properties of 2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one (ganciclovir) molecule is shown in Figure 1. The active conformation and electron density mapped of ganciclovir by ACDlabs-3D viewer software are shown in Figures 2 and 3 respectively. The highest occupied molecular orbital (HOMO) of ganciclovir molecule is shown in Figure 4. The lowest unoccupied molecular orbital (LUMO) of ganciclovir molecule is presented in Figure 5. Electrostatic potential of molecular ground state mapped onto the electron density surface of ganciclovir is presented in Figure 6. Self Consistent Field (SCF) energy graph of ganciclovir is shown in Figure 7. Atomic coordinates of ganciclovir molecule is given in Table 1. Bond length and bond angles are given in Tables 2 and 3 respectively, which are calculated after geometry optimization of molecule from Arguslab by using molecular mechanics calculation. Table 4 shows the Mulliken atomic charges and ZDO atomic charges of ganciclovir molecule.

Figure 1. Prospective view and calculated properties of 2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one(ganciclovir) molecule.
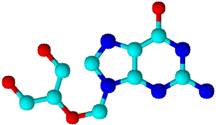
Figure 2. Active conformation of ganciclovir byACDlabs-3D viewer software.
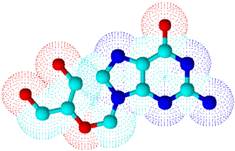
Figure 3. Electron density mapped of ganciclovir by ACDlabs-3D viewer software.
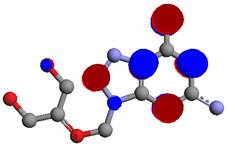
Figure 4. Highest Occupied Molecular orbitals (HOMO) of ganciclovir.
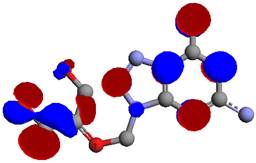
Figure 5. Lowest Unocupied Moleculars Orbitals (LUMO) of ganciclovir.
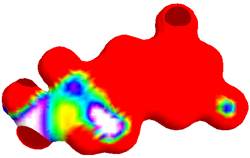
Figure 6. Electrostatic potential of ganciclovir mapped onto its electron density surface.
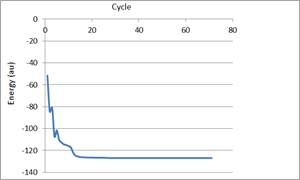
Figure 7. Self Consistent Field (SCF) energy ofGanciclovir.
Table 1. FinalAtomic Coordinates of ganciclovir.
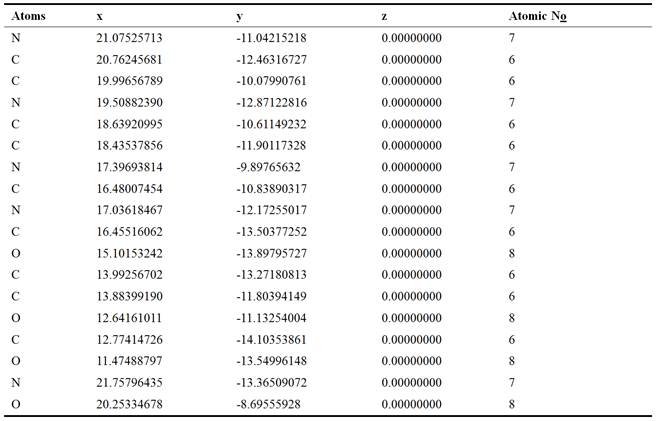
Table 2. Bond length of ganciclovir.
Table 3. Bond Angles of ganciclovir.
Tables 4. ZDO atomic charges and Mulliken atomic charges of ganciclovir.
4. Discussions
Chemical reactivity of a ganciclovir molecule depends on the interaction between HOMO (Figure 4) and LUMO (Figure 5) energy levels. The excited and ground states have different distributions of electron density. Frontier molecular orbitals predict fundamental principles that govern the reactivity of molecules. It predicts whether a molecule is a good nucleophile or a strong electrophile. It predicts which position of an aromatic ring undergoes electrophilic or nucleophilic substitutions. The simple understanding that a nucleophile is electron-rich molecule that attacks sites with low electron density, and that electrophile is electron-poor molecule that will attack a position of large electron density allows understanding many organic reactions [19]. A powerful practical model for describing chemical reactivity is the frontier molecular orbital (FMO) theory. The important aspect of the frontier electron theory is the focus on the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO). Instead of thinking about the total electron density in a nucleophile, we should think about the localization of the HOMO orbital because electrons from this orbital are most free to participate in reactions. Similarly, the frontier orbital theory predicts the site where the lowest unoccupied orbital is localized, which is a good electrophilic site [19]. The electrophilic site in ganciclovir molecule is represented in blue colour, while the nucleophilic site is show in red colour. The FMO theory can be used for explaining electrophilic substitution. The HOMO energy suggests the ability of the molecule to donate electrons to receptors. LUMO energy suggests the ability of the molecule to accept electrons.
The positive and negative phases (Figures 4 – 6) of the orbitalare represented by two colour, the red regions represent an increase in electron density and the blue regions shows a decrease in electron density. Electrostatic potential (ESP) surfaces of ganciclovir mapped onto the molecular electron density is shown in Figure 6. Electrostatic potential surfaces are valuable in computer-aided drug design because they help in optimization of electrostatic interactions between the protein and the ligand. These surfaces can be used to compare different inhibitors with substrates or transition states of the reaction [19]. What this surface simply shows is how the ESP has changed by the movement of electrons in this excitation. This surface gives a very direct indication of the shift of electron density. The blue end shows where the ESP has increased, which means where electron density has decreased. The red regions show where the ESP has decreased (become more negative) since the electron density has increased in this region. ESP at a point in space is the potential energy of a positive test charge at that point. If the electron density has decreased, then the interaction energy with the positive test charge will have become less negative, or increased, hence the blue color [9]. This type of surface representations is useful to discuss drug-receptor interaction. The colour map shows the electrostatic potential energy (in hartrees) for the various colours [9]. The red end of the spectrum shows regions of highest stability for a positive test charge, magenta/ blue show the regions of least stability for a positive test charge [9].
Mulliken and ZDO atomic charges have been applied to the estimation of reaction centres. It shows the charge distribution over the whole skeleton of the molecule [17]. It has been reported [17] that the more negatively charged heteroatoms have more tendency donate electrons to receptors. The calculated Mulliken and ZDO atomic charges of Ganciclovir (Table 4) shows numerous active centers. The site of receptors adsorption could be estimated from the net charges on a molecule.
Self consistent field energy (SCF) was obtained as ‘the minimum potential energy which is the needed energy for the interaction of drug with the receptor [16]. The SCF is the average interaction between a given particle and other particles of a quantum mechanical system consisting of many particles. Because the problem of many interacting particles is very complex and has no exact solution, calculations are done by approximate methods. One of the most often used approximated methods of quantum mechanics is based on the interaction of a self consistent field., which permits the many particle problem to be reduced to the problem of single particle moving in the average self consistent field produced by other particles [16]. The SCF energy (Figure 7) (Net Charge of -1, Valence electron of 86) was found to be -126.613168 au (-79451.034700 kcal/mol) as calculated by RHF/ AM1 method in ArgusLab 4.0.1 suite.
5. Conclusion
The present work indicates that the best conformation of 2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one (ganciclovir) was found to be -126.613168 au (-79451.034700 kcal/mol) as calculated by RHF/ AM1 method in ArgusLab 4.0.1 suite. This is the minimum potential energy calculated by ArgusLab 4.0.1software. At this point (2-amino-9-{[(1,3-dihydroxypropan-2-yl)oxy]methyl}-1,9-dihydro-6H-purin-6-one) (ganciclovir) will be more active to interact with the receptors. Such types of interactions are significant for drug- receptor interactions.
References
- Crumpacker, C.S.(1996). Ganciclovir. N Engl J Med, 335:721.
- Faulds, D. and Heel, R.C.(1990). Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections.Drugs 1990: 39:597.
- Markham, A. and Faulds, D.(1994). Ganciclovir. An update of its therapeutic use in cytomegalovirus infection.Drugs 1994: 48:455.
- Cohen, J.I., Davenport, D.S. and Stewart, J.A.(2002). Recommendations for prevention of and therapy forexposure to B virus (cercopithecine herpesvirus 1).Clin Infect Dis,35:1191.
- Mavros, M.N., Kapaskelis, A. and Falagas, M.E.(2010). Antiviral treatment for severe EBV infections in apparently immunocompetent patients.J Clin Virol,49:151.
- Venturi, C., Bueno, J. and Gavalda, J.(2009).Impact of valganciclovir on Epstein-Barr Virus polymerase chain reaction in pediatric liver transplantation: preliminary report.Transplant Proc, 41:1038.
- Crowder, G.A.(1986). Conformational analysis of 3,3-dimethylhexane, Int. J. Rapid. Comm.,19(7): 783–789.
- Cramer,C.J.(2004). Molecular Mechanics. In: Essentials of Computational Chemistry. Theories and Models, 2nd ed., John Wiley and Sons Ltd., England, 36.
- Thompson, M.(2004).ArgusLab 4.0.1.Planaria software LLC, Seattle.
- Dunn, I.I.I. and Hopfinger, AJ.(1998). Drug Discovery,Kluwer Academic Publishers, 12, 167.
- Cruciani, G., Clementi, S. and Pastor, M. (1998).Perspectives in DrugDiscovery and Design,16: 71.
- Dewar, M.J.S., Zoobisch, E.G., Healy, E.F. and Stewart, J.J.P. (1985). AM1: A new general purpose quantum mechanical molecular model, J. Am. Chem. Soc.107,3902–3910.
- Simons, J., Jorgensen, P., Taylor, H. and Ozment, J (1983) Walking on potential energy surfaces,J.Phys. Chem.87, 2745-2753.
- Csizmadia, I.G. and Enriz, R.D (2001). Peptide and protein folding,J. Mol.Struct.-Theochem.,543, 319–361.
- Martin, Y.C.(1998), Perspective in drug discovery and design, Springer Publisher, USA,12, 3-23.
- http://www.thefreedictionary.com
- Pearson, R.G(1986). Absolute Electronegativity and Hardness Correlated with Molecular Orbital Theory.Proceedings of the National Academy of Sciences of the United States of America, 83: 8440-8441.
- Rappe, A.K., Casewit, C.J., Colwell, K.S., Goddard III, W.A. and Skiff, W.M. (1992)."UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations",J. Am. Chem. Soc.,114:10024-10035.
- http://www.cmbi.ru.nl/molden/mapped.html
- Laxmi, K. (2014). Theoretical approach on structuralaspects of antiepileptic agent indoline-2,3-dione-3-oxime by arguslab 4 software. Journal of Applied Chemistry. 2(1): 92-101.
- Amaku, F. J, Otuokere, I. E and Igwe, K. K (2016). Computational study on the geometry optimization and excited – state properties of oxybuprocaine by ArgusLab 4.0.1 software, Journal of International Research in Medical and Pharmaceutical Sciences,8(1): 33-39.
- Amaku, F. J. and Otuokere, I. E. (2016)Theoretical Approach on Structural Aspects of a Potent, Selective, Orally Bioavailable Hedgehog Antagonist, 2-chloro-N-[4-chloro-3-(pyridin-2-yl)phenyl]-4-(methylsulfonyl)benzamide,Indian Journal of Advances in Chemical Science 4(1): 31-35
- Otuokere, I. E. and Alisa, C.O. (2014) Molecular mechanics steric energy evaluation of reversible acetyl cholinesterase inhibitor, donepezil, (RS)-2-([1-benzyl-4-piperidyl] methyl)-5,6-dimethoxy-2,3-dihydroinden-1-one, Indian Journal of Advances in Chemical Science, 3: 87-95.
- Amaku, F. J and Otuokere, I. E. (2015). Conformational Analysis of a Potent Anticancer Drug 3-(4-amino-1-oxo-1,3-dihydro-2h-isoindol-2-yl) Piperidine-2,6-Dione (Lenalidomide), International Journal of Materials Chemistry and Physic,1(3): 406–410.



