
American Journal of Educational Science, Vol. 1, No. 3, July 2015 Publish Date: Jun. 10, 2015 Pages: 47-51
Identify Unknown Bacteria with Restriction Enzyme and Its Related Factors
Tangjie Zhang*
College of Veterinary Medicine, Yangzhou University, Yangzhou, Jiangsu, China
Abstract
The plasmids used in molecular biology have been modified through genetic engineering to facilitate gene cloning and gene expression in bacteria. Cutting DNA and pasting DNA fragments together typically are among the first techniques learned in the molecular biology lab fundamental to all recombinant DNA work. To identify an unknown bacteria stock, restriction enzymes are used and the experimental conditions are discussed in this paper. The results shows that the unknown has 3 fragments of 1600bp, 1700bp and 2000bp and the evidence strongly suggested that the unknown bacteria is pAMP. Factors that influence restriction enzyme activity are also discussed.
Keywords
Restriction Enzyme, pAMP, Plasmid, Mini-Prep
Received:April 9, 2015
Accepted: May 9, 2015
Published online: June 8, 2015
@ 2015 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY-NC license. http://creativecommons.org/licenses/by-nc/4.0/
Contents
1. Introduction 2. Material and Methods 2.1. Perform Mini-Prep to Isolate Plasmid DNA from Unknown Bacteria Culture 2.2. Check the Quality of Plasmid DNA 2.3. Do the Restriction Digest Reaction Using the Enzyme EcoRV and Bsal 3. Results 4. Discussion 4.1. Restriction Enzymes 4.2. Factors That Influence Restriction Enzyme Activity 4.3. Mini-Prep Procedure to Isolate Plasmid DNA 5. Conclusion Acknowledgments
1. Introduction
Plasmids are circular pieces of DNA naturally found in bacterial cells. The plasmids used in molecular biology have been modified through genetic engineering to facilitate gene cloning and gene expression (protein production) in bacteria1-2. Antibiotic resistant genes have been engineered into these plasmids and function as selectable markers-that is to say, these genes allow us to select between bacteria that harbor the plasmid with those that do not. If a bacterium carries a plasmid with an antibiotic resistant gene, the bacterium will be able to grow and reproduce in the presence of that antibiotic; those bacteria without the plasmid will not be able to grow. Thus, antibiotics can be used to select bacteria that are resistant and presumably carry a plasmid with the resistant gene from those bacteria that do not carry the plasmid.
Our goal of this experiment is to figure out the identity of an unknown bacteria stock. Since restriction enzyme1, can recognize and cut DNA at specific sequence called restriction site, and each bacteria has their unique restriction sites, different bacteria will produce different number (if they have more than one restriction site) and sizes of DNA bands when treated with certain restriction enzyme. In this way, the unknown could be identified.
2. Material and Methods
2.1. Perform Mini-Prep to Isolate Plasmid DNA from Unknown Bacteria Culture
Pulse down cells in microcentrifuge on high (14,000 rpm) for 30 seconds. .Dump supernatent, suck off remaining culture, and resuspend cells in 100uL of ice cold Solution 1, vortexing ok. Add 200uL of freshly made Solution 2. Rock gently, incubate on ice until clear (~ 5 minutes). Add 150uL of ice cold Solution 3. Vortex immediately, incubate on ice ~ 5 minutes (For cleanest preps avoid transfering any precipitate). Microcentrifuge on high (14,000 rpm) for 10 minutes, remove supernatant to new microcentrifuge tubes (For cleanest preps avoid transfering any precipitate). Add 1mL of isopropanol to each tube (fill microcentrifuge tube). Cold isopropanol or incubation on ice may increase DNA precipitation yields (May be stored in freezer at this stage if necessary). Microcentrifuge on high (14,000 rpm) for 10 minutes, carefully discard supernatant, and add 1mL of 70% EtOH to wash pellet by gently mixing (Recentrifuge if pellet is disturbed for 5-10 min on high). Remove all supernatent with a pulled pasteur pippette, and allow remnants to evaporate on bench or in dessicator until pellet is just dry(Problems may be encountered drying to resuspend the pellet if allowed to overdry). Add 2uL of RNase A (10ug/mL) to 1 mL of TE buffer. Add 50uL of RNase A TE to pellet to resuspend, and incubate at 37 degrees for ~20 minutes.
Solution 1
50mM glucose
25mM Tris*Cl (pH 8.0)
10mM EDTA (pH 8.0)
Autoclave small batches of 100mL and store at 4 C.
Solution 2
0.1M NaOH (freshly diluted from 10M stock)
1% SDS
Solution 3
60mL 5M K*Acetate (Final Concentration 3M)
11.5mL Glacial Acetic Acid (Final Concentration 5M)
28.5mL dH2O
2.2. Check the Quality of Plasmid DNA
Use the plasmid DNA we got to perform gel electrophoresis in order to check the quality of plasmid DNA. We put 6µL plasmid DNA, 2 µL loading buffer and 4 µL dH2O in to a clean microcentrifuge tube. In another tube, prepare the molecular weight standard with 10 µL λDNA digested with HindIII and 2 µL of 6× loading dye. To do gel electrophoresis, we put a gel made of 0.8%(w/v) agarose into a buffer solution containing electrolytes, between a positive and negative electrode. We put our molecular weight standard and plasmid into 2 separate sample wells near the negative end. Since DNA will move to the positive end since they are negatively charged. Turn on the power and wait for the result.
If we get a major supercoiled band of dark color away from sample well, our plasmid is qualified for later use. The molecular weight standard will also help us roughly estimate the length of the DNA plasmid.
2.3. Do the Restriction Digest Reaction Using the Enzyme EcoRV and Bsal
In one microcentrifuge tube, we put 6µL plasmid, 2µL 5× loading buffer and 1µL EcoRV and 1µL Bsal in sequence. The plasmid is going to be cut by these two enzyme. In another tube, we prepare our negative control by adding 6µL plasmid, 2µL 5× loading buffer and 2 µL sterile water. These are going to produce an uncut DNA. We use a commercially prepared HindIII/EcoRI digest as molecular weight standard. Prepare the gel as in step2 and load these to three separate sample wells. Turn on the electric power.
If the unknown is pAMP, there will be 3 bands of 3459-2096=1363 base pairs, 2096-417=1679 base pairs and 4539-1363-1679=1497 base pairs. If the unknown is pKAN, there will be only one band of 4194 base pair. If the unknown is pGLO, there will be two bands, 3357-386=2971 base pair and 5371-2971=2400 base pair. These values are based on the restriction site of EcoRV and Bsal on pAMP, pKAN and pGLO we know. The three plasmids used in the lab have been modified from two commercially available plasmids, pGLO™ (Bio-Rad), pAMPand pKAN (DNA Learning Center at Cold Spring Harbor Laboratory).
3. Results
Table1 shows the relationship between moving distance of 12 different fragments and their respective sizes, produced by our molecular weight standard (cut by HindIII and EcoRI).
Table 1. The relative mobility of standard.
| Fragment size | Relative mobility |
| 21226 | 5 |
| 5148 | 6 |
| 4973 | 6.2 |
| 4268 | 6.6 |
| 3530 | 7.1 |
| 2027 | 8.2 |
| 1904 | 8.3 |
| 1584 | 8.8 |
| 1375 | 9.2 |
| 947 | 10.2 |
| 831 | 10.5 |
| 564 | 11.6 |
Then, we show these values on graph and connected them by a logarithmic line (Figure1), in order to see the relationship of relative mobility and fragment size, so that we can later predict the fragment size of our unknown.
We use HindIII digest of λDNA as molecular weight standard, and let it run on our gel. The DNA was cut into many fragments of different size. We then find their corresponding fragment and measure their relative mobility.
When we measure our unknown’s moving distance on the gel, we see 3 different fragments moving 8.4cm, 8.7cm, and 8.9cm away from the negative end.
We use the trend line above and determine the size of these fragments, which are about 1600 base pair, 1700 base pairs and 2000 base pairs.
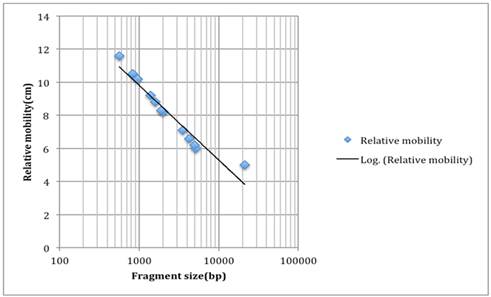
Figure 1. The Moving Distance of Molecular weight standard-Relative mobility v.s. Fragment Size.
4. Discussion
Plasmids are circular pieces of DNA that are naturally found in bacterial cells.
Virtually all bacterial species plasmids exist. These accessory genetic elements typically account for only a small fraction of a bacterial genome corresponding to a range between 1 and 200 kb. Extremely large plasmids with sizes far beyond 200 kb are also known. Plasmids of more than 50 kb might be characterized as "large", and plasmids of less than 10 kb as small".
4.1. Restriction Enzymes
Restriction enzymes play an essential role in recombinant DNA technology. The significance of these enzymes cannot be overstated since the biotechnology industry has been built upon these enzymes to a large extent.
Prior to the discovery of restriction enzymes, it was observed that certain strains of bacteria seemed to be immune to bacteriophages3. These bacteria were producing an unknown molecular "agent" that appeared to function as a primitive immune system, which restricted the growth of viruses. In the early 1970’s, Hamilton Smith and Daniel Nathans were able to purify some of these immune agents4. They were discovered to be enzymes that bacteriums produced and used to cut-up the viral DNA as it was injected into cells. When the virus introduced its DNA into the bacterial cell, the enzymes cleaved the viral DNA into fragments, thus preventing or restricting the growth of the virus.
There are several classes of restriction enzymes, but the ones that have been most useful are the specific endonucleases. These endonucleases cut the DNA molecule internally, not from the ends as exonucleases will cut. Because many of these endonucleases are specific, they consistently recognize a specific nucleotide base sequence, the recognition sequence. Some recognize a four-base sequence; others recognize a five or six-base sequence. The important feature is that a given restriction endonuclease will always recognize the same nucleotide sequence and cut the DNA at its restriction site. The recognition sequences are palindromes. Some restriction enzymes will make a "blunt cut," leaving no overhanging bases. Other enzymes will leave overhanging bases creating "sticky ends." These enzymes are particularly useful; since sticky ends make recombining DNA fragments a fairly simple procedure. The "stickiness" is the result of the extraordinary affinity of complimentary nucleotides to form hydrogen bonds between them.
4.2. Factors That Influence Restriction Enzyme Activity
It is not uncommon to have difficulties in digesting DNA with restriction enzymes. At times, the DNA does not appear to be cut at all, or sometimes it is only partially cut. Restriction sites can be predicted if the sequence is known. However, an enzyme may cut more often than it should or at the wrong sites in labs due to a variety of factors. In some cases, these unexpected results were not caused by problems in techniques: for example, the sequence you have may be incorrect, or a restriction map provided by a colleague could be in error. However, there are a number of commonly-encountered situations that influence how well restriction enzymes cut, and it is important to be aware of these for troubleshooting.
In an experiment like this, the following factors might influence restriction enzyme activity that we should take into consideration.
4.2.1. Incubation Temperature
Most restriction enzymes cut best at 37℃, but there are many exceptions. Enzymes isolated from thermophilic bacteria cut best at temperatures ranging from 50 to 65℃. Some other enzymes have a very short half life at 37℃ and it is recommended that they are incubated at 25℃.
4.2.2. Star Activity
When DNA is digested with certain restriction enzymes under non-standard conditions, cleavage can occur at sites different from the normal recognition sequence - such aberrent cutting is called "star activity". An example of an enzyme that can exhibit star activity is EcoRI; in this case, cleavage can occur within a number of sequences that differ from the canonical GAATTC by a single base substitution.
4.2.3. Buffer Composition
Different restriction enzymes have differing preferences for ionic strength (salt concentration) and major cation (sodium or potassium). A battery of 3 to 4 different buffers will handle a large number of available enzymes, although there are a few enzymes that require a unique buffer environment. A major function of the buffer is to maintain pH of the reaction (usually at 8.0). Additionally, some enzymes are more fussy about having their optimal buffer than other enzymes. Clearly, use of the wrong buffer can lead to poor cleavage rates.
4.2.4. DNA Methylation
Almost all strains of E. coli bacteria used for propagating cloned DNA contain two site-specific DNA methylases:
Dam methylase adds a methyl group to the adenine in the sequence GATC, yielding a sequence symbolized as GmATC.
Dcm methylase methylates the internal cytosine in CC(A/T)GG, generating the sequence CmC(A/T)GG.
The practical importance of this phenomenon is that a number of restriction endonucleases will not cleave methylated DNA5-6. The take-home message here is that if DNA unexpectedly does not cut or cuts only partially, check that the enzyme in question is not methylation-sensitive.
4.2.5. Digestion with Multiple Enzymes
Digesting DNA with two enzymes is a commonplace task, and oftentimes the two enzymes have different buffer requirements. There are at least three ways to handle this situation:
Digest with both enzymes in the same buffer. In many cases, even though a given buffer is not optimal for an enzyme, you can still get quite good cleavage rates. Enzyme manufacturer catalogs usually contain a reference table recommended the best single buffer for conducting specific double digests.
Cut with one enzyme, then alter the buffer composition and cut with the second enzyme. This usually applies to situations where one enzyme like a low salt buffer and the other a high salt buffer, in which case you can digest with the first enzyme for a time, add a calculated amount of concentrated NaCl and cut with the second enzyme.
Change buffers between digestion with two enzymes. In some cases, two enzymes will have totally incompatible buffers. In that case, perform one digestion, recover the DNA (usually by precipitation) and resuspend in the buffer appropriate for the second enzyme.
4.2.6. Variability in Digestion of Different DNA Substrates
The efficiency with which a restriction enzyme cuts its recognition sequence at different locations in a piece of DNA can vary 10 to 50-fold. This is apparently due to influences of sequences bordering the recognition site, which perhaps can either enhance or inhibit enzyme binding or activity.
A related situation is seen when restriction recognition sites are located at or very close to the ends of linear fragments of DNA. Most enzymes require a few bases on either side of their recognition site in order to bind and cleave. Many of the companies that sell enzymes provide a table in their catalog that presents "end requirements" for a variety of enzymes.
4.3. Mini-Prep Procedure to Isolate Plasmid DNA
Mini-Prep procedure is used to isolate small plasmid DNA from bacteria while limiting contaminating proteins and genomic DNA. The plasmid quality is acceptable for restriction analysis, sequencing, cloning, or other purposes. Minipreparation of plasmid DNA is a rapid, small-scale isolation of plasmid DNA from bacteria. It is based on the alkaline lysis method invented by the researchers Birnbaum and Doly in 19797. The extracted plasmid DNA resulting from performing a miniprep is itself often called a "miniprep". Minipreps are used in the process of molecular cloning to analyze bacterial clones8-9. A typical plasmid DNA yield of a miniprep is 50 to 100 µg depending on the cell strain. Miniprep of large number of plasmids can also be done conveniently by filter paper lysing, the elution of the filter paper that contains plasmid can be directly sequenced to produce more than 700 bp high quality sequencing data with CE sequencing10-11.
The procedures are based on the fact that plasmids usually occur in the covalently closed circular (supercoiled) ccc configuration within the host cells. After gentle cell lysis all intracellular macromolecules have to be eliminated whereas plasmid DNA is enriched and purified. The smaller a plasmid, the easier the isolation of intact ccc molecules is. DNA is very sensitive to mechanical stress, therefore shearing forces caused by mixing/vortexing or fast pipetting must be avoided as soon as cell lysis occurs. All mixing steps during and after cell
lysis should be performed carefully by inverting the tubes several times (8-10 fold). Especially in case of larger plasmids it is recommended to cut off the ends of plastic pipette tips to minimize shearing forces. Gloves should be worn in order to prevent contamination with DNases. Autoclaved (DNase-free) buffer solutions, tubes and tips should be used. If phenotypic markers of a plasmid (e.g. antibiotic resistances) are known, it is recommended to grow the cells under selective pressure to avoid plasmid loss. If necessary, small plasmids of Escherichia coli can easily be amplified using chloramphenicol. This results in several thousand plasmid copies per cell leading to high DNA quantities (Clewell, 1972). Large plasmids are maintained with only one copy per host chromosome: visible DNA bands are more difficult to get. For plasmid isolation, bacterial cultures should be grown to late logarithmic/early stationary phase. It is important to remove the supernatant completely after centrifugation from the cell pellets. Tris buffer is the typical buffering substance for DNA with buffering capacity in the slightly alkaline range in which DNA can also be stored best (pH 7.5-8.2). EDTA is an important substance in plasmid preparations because it inhibits nuclease activity. For long-term storage, plasmid DNA should be frozen in aliquots of storage TE buffer. Repeated thawing and freezing of DNA should be avoided.
5. Conclusion
Based on our results that the unknown has 3 fragments of 1600bp, 1700bp and 2000bp, we find the unknown to be pAMP when compared with our prediction. Even though the actual fragment size is little bigger than our expectation, the evidence still strongly proved that the unknown is pAMP. Incubation temperature, star activity, buffer composition, DNA methylation, multiple enzymes and variability in digestion of different DNA substrates could affect restriction enzyme activity. Mini-Prep procedure is suggested to isolate small plasmid DNA from bacteria while limiting contaminating proteins and genomic DNA.
Acknowledgments
This research was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions and Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou, 225009.
References
- Scott Freeman et al.. Biological Science,5thedition, 2013,p 370-371.
- Birnboim, H.C. and Doly, J. A rapid alkaline procedure for screening recombinant plasmid DNA[J]. Nucleic Acids Res. 1979,7: 1513–1523.
- Lenski R E. Experimental studies of pleiotropy and epistasis in Escherichia coli. I. Variation in competitive fitness among mutants resistant to virus T4[J]. Evolution, 1988: 425-432.
- Loenen W A M, Dryden D T F, Raleigh E A, et al. Highlights of the DNA cutters: a short history of the restriction enzymes[J]. Nucleic acids research, 2014, 42(1): 3-19.
- Tahiliani M, Koh K P, Shen Y, et al.Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1[J]. Science, 2009, 324(5929): 930-935.
- McClelland M. The effect of sequence specific DNA methylation on restriction endonuclease cleavage[J]. Nucleic acids research, 1981, 9(22): 5859-5866.
- Birnboim HC, Doly J (November 1979). "A rapid alkaline extraction procedure for screening recombinant plasmid DNA". Nucleic Acids Res. 7 (6): 1513–23.
- Sambrook, J., Russell, D.W., editors. Molecular Cloning, A Laboratory Manual. 3rd ed. Cold Spring Harbor Laboratory Press, New York, 2001
- Tartof, K.D., Hobbs, C. A. 1987. Improved media for growing plasmid and cosmid clones. Bethesda Res. Lab. Focus 9, 12.
- Lipps, G., editor. Plasmids – Current Research and Future Trends. Caister Academic Press, Norfolk, UK. 2008.
- Meyers, J.A., Sanchez, D., Elwell, L.P., Falkow, S. 1976. Simple agarose gelelectrophoretic method for the identification and characterization of plasmiddeoxyribonucleic acid. J. Bacteriol. 127, 1529-1537.



