
American Journal of Clinical Neurology and Neurosurgery, Vol. 2, No. 4, July 2016 Publish Date: Aug. 19, 2016 Pages: 45-50
Neurogenetic Disorders of Down Syndrome and Potential Pharmacotherapies for Mental Retardation
Mohammed Rachidi*
Molecular Genetics of Human Diseases, French Polynesia; University Paris 7 Denis Diderot, Paris, France
Abstract
Background: Trisomy of human chromosome 21 is the most frequent genetic cause of mental retardation or intellectual disability and others phenotypes, including developmental defects, dysmorphic features and cognitive impairments collectively known as Down syndrome. Mainly a consequence of developmental and functional brain alterations, the mental retardation is the most invariable and invalidating neuropathological characteristic caused by the overdosage of genes triplicated in the chromosome 21. Methods and Results: The cytogenetic and molecular analysis facilitate the identification of the minimal region or Down Syndrome Chromosomal Region responsible for many phenotypes including mental retardation as the major constant phenotype caused by the overexpression of chromosome 21 genes. The complete sequence of human chromosome 21 and the transcriptome analysis in Down syndrome patients and in trisomic mouse models facilitate the genetic dissection of neurological and cognitive phenotypes. As a result of high degree of conservation of genomes and of molecular mechanisms between mouse and human, the mouse models of Down syndrome showed similar neuropathological features seen in Down syndrome persons and facilitate the identification of associated genetic targets. Conclusion: The genetic dissection of neurological phenotypes in trisomic mouse models highly developed our understanding of cellular and molecular mechanisms of gene overexpression caused by trisomy 21 and contributed significantly to the identification of specific genetic targets for pharmacological therapeutics. These pharmacological treatments in mouse models of Down syndrome allowed successfully post-drug rescue of neurological alterations and associated cognitive deficits and could be useful therapeutic tools of neurocognitive deficits and mental retardation seen in Down syndrome persons.
Keywords
Down Syndrome and Trisomy 21, Mental Retardation, Trisomic Mouse Models, Neurological Phenotypes,
Learning and Memory, Molecular Targets, Genetic Pathways, Pharmacotherapies.
Received:January 11, 2016
Accepted: May 3, 2016
Published online: August 19, 2016
@ 2016 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY license. http://creativecommons.org/licenses/by/4.0/
Contents
1. Introduction
Trisomy of human chromosome 21 (HSA21) or Down syndrome (DS), affecting 1 in 700 newborns, produces a variety of developmental anomalies in different organs involving dysmorphic features, hypotonia, immunological, haematological and endocrinal defects, neurological and neurotransmitter alterations, and increased occurrence of Alzheimer’s disease [1-3]. DS is also a risk factor for a number of diseases, such as cardiac malformations, childhood onset leukemia and Hirschsprung disease [4-7].
Down syndrome is the most frequent genetic cause of mental retardation or intellectual disability that is mainly a consequence of functional and developmental brain alterations. DS individuals are characterized by several neurological defects in cortex lamination, in the shape and volume of several cerebral and cerebellar regions, cognitive impairments and low Intelligence Quotient (IQ). The brain morphological alterations have been found at cellular level determined by alteration in neurogenesis, neuronal differentiation, myelination, dendritogenesis and synaptogenesis [8-12].
The genetic discovery of DS genetic and neurocognitive phenotypes in trisomic mouse models highly developed our understanding of cellular and molecular mechanisms of gene overexpression caused by trisomy of human chromosome 21 and contributed to the discovery of associated genetic pathways and molecular targets for potential pharmacotherapies.
2. Genetic and Neurocognitive Disorders
Generally, DS results from an extra copy of chromosome 21 in all cells of the afflicted individuals and in some rare cases DS results from a partial trisomy 21 showing variable phenotypes depending of the triplicated region. The cytogenetic and molecular analysis of such patients allowed the discovery of the minimal region or Down Syndrome Chromosomal Region (DSCR), at 21q22.2 sub-band, responsible for many traits of DS, including mental retardation as the major constant phenotype caused by the overdosage of genes triplicated in the chromosome 21 [13,14]. The complete sequence of human chromosome 21 [15], the sequence of the human genome [16], and the transcriptome investigations in DS patients and in trisomic mouse models [17-20] facilitate the genetic dissection of DS neurological and cognitive phenotypes.
The Ts(1716)65Dn, named Ts65Dn, produced by reciprocal translocation T(16C3-4; 17A2)65Dn, is the first segmental trisomic mice created and is the most commonly used and the best-characterized DS mouse models [21]. Ts65Dn mice are trisomic for the most of HSA21 orthologous genes conserved in the distal end of mouse chromosome 16 and exhibit many neurological features that are reminiscent of those seen in people with DS. The Ts65Dn mouse models showed neurotransmitter alterations [22], reduced hippocampal volume and dentate gyrus density [23], decreased Long-Term Potentiation (LTP) and increased Long-Term Depression (LTD) in the brain [24,25], reduced cerebellum and granular cell layers [26], degeneration of Basal Forebrain Cholinergic Neurons (BFCN) [27,28], decreased synaptic density in the cortex [29], decreased neuronal density and decreased synaptic density in the dentate gyrus [30], enlarged synapses in the cortex [31]. The abnormal cognitive behaviors, analogous to DS cognitive defects, have been demonstrated using different behavioral tests for spatial learning and memory such as T-maze, Y-maze, radial maze and Morris water maze. The Ts65Dn mouse models of DS showed motor dysfunction [32], reduced responsiveness to painful stimuli [33], decreased fear conditioning [34], decreased spatial learning and memory in the Morris water maze, the radial-arm maze, the water T-maze and the water radial-arm maze [35-39].
Furthermore, the mouse models of DS are powerful tools that greatly enhanced our understanding of the molecular and cellular mechanisms involved in DS pathogenesis because of the possibility to genetically manipulate their genome, the tissue accessibility and the high degree of conservation of genomes and molecular mechanisms between mouse and human. Interestingly, these mouse models demonstrate similar DS neurological and cognitive phenotypes and have significantly contributed to the discovery of altered genetic pathways associated to DS neurological and cognitive features [40]. Remarkably, an altered genetic pathway implicated in some neurological and cognitive DS phenotypes has been identified in which two critical HSA21 genes are involved and located in the Down syndrome critical region (DSCR). MNB / DYRK1A (Minibrain / Dual specificity tyrosine phosphorylation-Regulated Kinase 1A) and DSCR1 or RCAN1 (Regulator of the Calcineurin 1 protein) operate synergistically to control the phosphorylation levels of Nuclear Factor of Activated T cells (NFATc) and NFATc-regulated gene expression [41]. It has been demonstrated that the overexpression of DYRK1A and RCAN1 genes dysregulates the NFATc pathway which play an essential role in the central nervous system and that the NFATc mice show neurological dysfunctions similar to those seen in DS patients and also in the Ts65Dn mice, the famous and the most extensively studied trisomic mouse models of DS [41].
3. Genetic Targets and Pharmacotherapies
The mouse models overexpressing DYRK1A gene show a significant impairment in spatial learning and memory in the behavioral tests, indicating hippocampal and prefrontal cortex function alterations, particularly concerning a cognitive dysfunction of the reference memory. Moreover, these transgenic mice show abnormal long-term potentiation (LTP) and abnormal long-term depression (LTD), suggesting also a synaptic plasticity alteration [42-44]. These functional brain alterations are comparable with those found in trisomic mouse models of DS and suggest a causative role of DYRK1A in mental retardation in DS persons.
Interestingly, the treatment of DYRK1A transgenic mice with injection into striatum of inhibitory Dyrk1A shRNA restores the motor coordination, attenuates the hyperactivity and improves the sensorimotor gating [45]. In addition, the treatment of DYRK1A transgenic mice with epigalloctechin-3-gallate, a major polyphenolic component of green tea, rescues brain alterations and improve cognitive deficits induced by the overexpression of DYRK1A gene [46] indicating DYRK1A as a therapeutic target (TABLE 1).
Table 1. Treatments and pharmacotherapeutic effects in mouse models of Down syndrome.
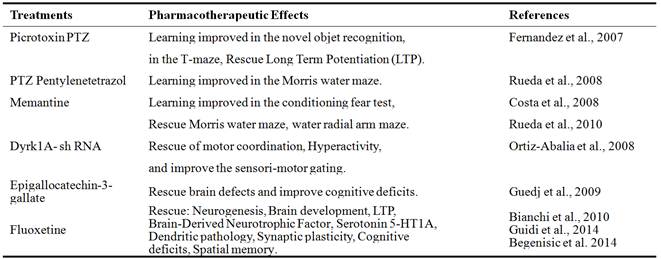
Some pharmacotherapies for cognitive impairments in trisomic mouse models of Down syndrome have been developed. It has been demonstrated that administering the GABAAantagonistspicrotoxin, bilobalide or Pentylenetetrazol (PTZ) restored cognition and long-term potentiation LTP in the Ts65Dn mouse models of DS [47]. These studies were confirmed using the non-competitive GABAA antagonist Pentylenetetrazole (PTZ) that rescued learning and memory performances of Ts65Dn trisomic mice in the Morris water maze tests [48]. These findings illustrate that GABAergic inhibition of specific brain circuits is a potential cause of intellectual disability in DS, and that GABAA antagonists may be useful therapeutic tools to facilitate the functional changes that can improve the cognitive deficits (TABLE 1).
In other similar experiments, it has also been established that acute injections of the N-methyl-D-aspartate (NMDA) receptor antagonist memantine rescue the performance deficits in the Ts65Dn mouse models of DS on a conditioning fear test, and that one target of memantine is the NMDA receptor, whose function is predicted to be disturbed by the integrated effects of increased expression of critical HSA21 genes RCAN1 and DYRK1A [49,50]. Remarkably, these treatments with NMDA receptor or GABAAantagonists allowed post-drug rescue of cognitive deficits in mouse models of DS indicating a hopeful post-drug rescue of neurocognitive deficits seen in DS persons.
More importantly, other pharmacological approaches have been developed recently to treat mouse models of DS in utero and demonstrated therapeutic effects that persisted to adulthood. In the Ts65Dn trisomic mice, fluoxetine treatment restored the expression of serotonin 5-HT1A receptors and
Brain-Derived Neurotrophic Factor (BDNF) and rescued also the cognitive deficits [51]. The early treatment of Ts65Dn trisomic mice with fluoxetine fully restored all the defects of the dendritic pathology (hypotrophic dendritic arbor, fewer spines and reduced innervations) in the dentate gyrus [52]. In adulthood, it has been demonstrated also that fluoxetine normalizes GABA release and rescues hippocampal synaptic plasticity and spatial memory in trisomic mouse models of DS [53].
These early treatments in mouse models of DS are of the most interest because they allowed successfully and completely post-drug rescue of neurological alterations and associated cognitive deficits and could be useful therapeutic tools of neurocognitive deficits and mental retardation seen in DS persons.
4. Conclusion
In parallel to development of genome whole technologies and the promising experimental results obtained in mouse models of DS, considerable progresses have been made in the last years in the cellular biology and molecular genetics of DS. The genetic investigations of DS neurological phenotypes in trisomic mouse models highly developed our understanding of cellular and molecular mechanisms of gene overexpression caused by trisomy 21 and the related molecular pathways involved in the functional neurogenetic and associated cognitive disorders seen in DS persons. Remarkably, these advances contributed significantly to the identification of specific genetic targets for pharmacological therapeutics of neurocognitive impairments in Down syndrome, particularly the mental retardation.
References
- Lejeune, J. (1990). Pathogenesis of mental deficiency in trisomy 21. American Journal Med. Genetics, 7 (Suppl.): 20-30.
- Stoll, C., Alembik, Y., Dott, B. and Roth, M.P. (1990). Epidemiology of Down syndrome in 118,265 consecutive births. American Journal Med. Genetics, 7 (Suppl.): 79-83.
- Pulsifer, M.B. (1996). The neuropsychology of mental retardation. J. Int. Neuropsychol. Soc., 2: 159-176.
- Ferencz, C., Neill, C.A., Boughman, J.A., Rubin, J.D., Brenner, J.I. and Perry, L.W. (1989). Congenital cardiovascular malformations associated with chromosome abnormalities: An epidemiologic study. Journal Pediatr., 114: 79-86.
- Puffenberger, E.G., Kauffman, E.R., Bolk, S., Matise, T.C., Washington, S.S., Angrist, M., Weissenbach, J., Garver, K.L., Mascari, M. and Ladda, R. (1994). Identity-by-descent and association mapping of a recessive gene for Hirschsprung disease on human chromosome 13q22. Human Mol. Genet., 3: 1217-1225.
- Epstein, C.J. (1995). Down syndrome (trisomy 21). In: Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D. (Eds.), The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill Inc., New York, pp. 749–794.
- Cohen,W.I. (1999). Health care guidelines for individuals with Down syndrome: 1999 revision. Down Synd. Quart., 4: 1-16.
- Wisniewski, K.E. (1990). Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. American Journal Med. Genetics., 7: 274-281.
- Raz, N., Torres, I.J., Briggs, S.D., Spencer, W.D., Thornton, A.E., Loken,W.J., Gunning, F.M., McQuain, J.D., Driesen, N.R. and Acker, J.D. (1995). Selective neuroanatomic abnormalities in Down’s syndrome and their cognitive correlates: evidence from MRI morphometry. Neurology, 45: 356-366.
- Hodapp, R.M., Ewans, D.E. and Gray, F.L. (1999). Intellectual development in children with Down syndrome. In: Rondal, J.A., Perera, J., Nadel, L. (Eds.), Down Syndrome: A Review of Current Knowledge. Whurr Publisher, London, pp. 124-132.
- Chapman, R.S. and Hesketh, L.J. (2000). Behavioral phenotype of individuals with Down syndrome. Mental Retardation Developmental Disabilities. Research. Rev., 6: 84-95.
- Brown, J.H., Johnson, M.H., Patterson, S.J., Gilmore, R., Longhi, E., and Karmiloff-Smith, A. (2003). Spatial representation and attention in toddlers with Williams syndrome and Down syndrome. Neuropsychologia, 41: 1037-1046.
- Rahmani, Z., Blouin, J.L., Creau-Goldberg, N., Watkins, P.C., Mattei, J.F., Poissonnier, M., Prieur, M., Chettouh, Z., Nicole, A., Aurias, A., Sinet, P.M. and Delabar, J.M. (1989).Critical role of the D21S55 region on chromosome 21 in the pathogenesis of Down syndrome. Proc. Natl. Acad. Sci., 86: 5958-5962.
- Delabar, J.M., Theophile, D., Rahmani, Z., Chettouh, Z., Blouin, J. L., Prieur, M., Noel, B. and Sinet, P. M. (1993). Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur. J. Human Genetics, 1: 114-124.
- Hattori, M., Fujiyama, A., Taylor, T.D., Watanabe, H., Yada, T., Park, H.S., Toyoda, A., Ishii, K., Totoki,Y., Choi, D.K., Groner, Y., Soeda, E., Ohki, M., Takagi, T., Sakaki, Y., Taudien, S., Blechschmidt, K., Polley, A., Menzel, U., Delabar, J., Kumpf, K., Lehmann, R., Patterson, D., Reichwald, K., Rump, A., Schillhabel, M., Schudy, A., Zimmermann,W., Rosenthal, A., Kudoh, J., Schibuya, K., Kawasaki, K., Asakawa, S., Shintani, A., Sasaki, T., Nagamine, K., Mitsuyama, S., Antonarakis, S.E., Minoshima, S., Shimizu, N., Nordsiek, G., Hornischer, K., Brant, P., Scharfe, M., Schon, O., Desario, A., Reichelt, J., Kauer, G., Blocker, H., Ramser, J., Beck, A., Klages, S., Hennig, S., Riesselmann, L., Dagand, E., Haaf, T.,Wehrmeyer, S., Borzym, K., Gardiner, K., Nizetic, D., Francis, F., Lehrach, H., Reinhardt, R. and Yaspo, M.L., (2000). The DNA sequence of human chromosome 21. Nature, 405: 311-319.
- International Human Genome Sequencing Consortium (2004). Finishing the euchromatic sequence of the human genome. Nature, 431: 931-945.
- Mao, R., Zielke, C. L., Zielke, H. R. and Pevsner, J. (2003). Global up-regulation of chromosome 21 gene expression in the developing Down syndrome brain. Genomics, 81: 457-467.
- Lyle, R., Gehrig, C., Neergaard-Henrichsen, C., Deutsch, S. and Antonarakis, S.E. (2004). Gene expression from the aneuploid chromosome in a trisomy mouse model of Down syndrome. Genome Res., 14: 1268-1274.
- Deutsch, S., Lyle, R., Dermitzakis, E.T., Attar, H., Subrahmanyan, L., Gehrig, C., Parand, L., Gagnebin, M., Rougemont, J., Jongeneel, C.V. and Antonarakis, S.E. (2005). Gene expression variation and expression quantitative trait mapping of human chromosome 21 genes. Human Molecular Genetics, 14: 3741-3749.
- Sultan, M., Piccini, I., Balzereit, D., Herwig, R., Saran, N.G., Lehrach, H., Reeves, R. H. and Yaspo, M. L. (2007). Gene expression variation in Down's syndrome mice allows prioritization of candidate genes. Genome Biol., 8: R91.
- Davisson, M.T., Schmidt, C. and Akeson, EC. (1990). Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog. Clin. Biol. Res., 360: 263-280.
- Dierssen, M., Vallina, I.F., Baamonde, C., Garcia-Calatayud, S., Lumbreras, M.A. and Florez, J. (1997).Alterations of central noradrenergic transmission in Ts65Dn mouse, a model for Down syndrome. Brain Res., 749: 238-244.
- Insausti, A.M., Megias, M., Crespo, D., Cruz-Orive, L.M., Dierssen, M., Vallina, I.F., Insausti, R., Florez, J. and Vallina, T.F. (1998). Hippocampal volume and neuronal number in Ts65Dn mice: a murine model of Down syndrome. Neuroscience Lett., 253: 175-178.
- Siarey, R.J., Stoll, S.I., Rapoport, S.I. and Galdzicki, Z. (1997). Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down syndrome. Neuropharmacology, 36: 1549-1554.
- Siarey, R.J., Carlson, E.J., Epstein, C.J., Balbo, A., Rapoport, S.I. and Galdzicki, Z. (1999). Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology, 38: 1917-1920.
- Baxter, L.L., Moran, T.H., Richtsmeier, J.T., Troncoso, J. and Reeves, R.H. (2000). Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Human Mol. Genet., 9: 195-202.
- Granholm, A.C., Sanders, L.A. and Crnic, L.S. (2000). Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp. Neurol., 161: 647-663.
- Salehi, A., Delcroix, J.D., Belichenko, P.V., Zhan, K., Wu, C., Valletta, J.S., Takimoto-Kimura, R., Kleschevnikov, A.M., Sambamurti, K., Chung, P.P., Xia,W., Villar, A., Campbell,W.A., Kulnane, L.S., Nixon, R.A., Lamb, B.T., Epstein, C.J., Stokin, G.B., Goldstein, L.S. and Mobley, W.C. (2006). Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron, 51: 29-42.
- Kurt, M.A., Davies, D.C., Kidd, M., Dierssen, M. and Florez, J. (2000). Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res., 858: 191-197.
- Kurt, M.A., Kafa, I.M., Dierssen, M. and Davies, C.D. (2004). Deficits of neuronal density in CA1 and synaptic density in the dentatgyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Res., 1022: 101-109.
- Belichenko, P.V., Masliah, E., Kleschevnikov, A.M., Villar, A.J., Epstein, C.J., Salehi, A. and Mobley, W.C. (2004). Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol., 480: 281-298.
- Costa, A.C., Walsh, K. and Davisson, M.T. (1999). Motor dysfunction in a mouse model for Down syndrome. Physiol. Behav., 68: 211-220.
- Martinez-Cue, C., Baamonde, C., Lumbreras, M.A., Vallina, I.F., Dierssen, M. and Florez, J. (1999). A murine model for Down syndrome shows reduced responsiveness to pain. Neuroreport, 10: 1119-1122.
- Hyde, L.A., Frisone, D.F., Crnic, L.S., 2001. Ts65Dn mice, a model for Down syndrome, have deficits in context discrimination learning suggesting impaired hippocampal function. Behav. Brain Res. 118, 53-60.
- Reeves, R.H., Irving, N.G., Moran, T.H., Wohn, A., Kitt, C., Sisodia, S.S., Schmidt, C., Bronson, R.T. and Davisson, M.T. (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics, 11: 177-184.
- Demas, G.E., Nelson, R.J., Krueger, B.K. and Yarowsky, P.J. (1996). Spatial memory deficits in segmental trisomic Ts65Dn mice. Behav. Brain Res., 82: 85-92.
- Demas, G.E., Nelson, R.J., Krueger, B.K. and Yarowsky, P.J. (1998). Impaired spatial working and reference memory in segmental trisomy (Ts65Dn) mice. Behav. Brain Res., 90: 199-201.
- Sago, H., Carlson, E.J., Smith, D.J., Rubin, E.M., Crnic, L.S., Huang, T.-T. and Epstein, C.J. (2000). Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatr. Res., 48: 606-613.
- Bimonte-Nelson, H.A., Hunter, C.L., Nelson, M.E. and Granholm, A.C. (2003). Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behav. Brain Res., 139: 47-57.
- Rachidi, M. and Lopes, C. (2008).Mental retardation and associated neurological dysfunctions in Down syndrome: a consequence of dysregulation in critical chromosome 21 genes and associated molecular pathways. Eur. J. Paediatric Neurology,12: 168-182.
- Arron, J. R., Winslow, M. M., Polleri, A., Chang, C. P., Wu, H., Gao, X., Neilson, J. R., Chen, L., Heit, J. J., Kim, S. K., Yamasaki, N., Miyakawa, T., Francke, U., Graef, I. A. and Crabtree, G. R., (2006). NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature,441:595-600.
- Smith, D.J., Stevens, M.E., Sudanagunta, S.P., Bronson, R.T., Makhinson, M.Watabe, A.M., O’Dell, T.J., Fung, J.,Weier, H.U., Cheng, J.F. and Rubin, E.M. (1997). Functional screening of 2 Mb of human chromosome 21q22,2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nature Genetics, 16, 28-36.
- Altafaj, X., Dierssen, M., Baamonde, C., Marti, E., Visa, J., Guimera, J., Oset, M., Gonzalez, J.R., Florez, J., Fillat, C. and Estivill, X. (2001).Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Human Molecular Genetics, 10: 1915-1923.
- Ahn, K.J., Jeong, H.K., Choi, H.S., Ryoo, S.R., Kim, Y.J., Goo, J.S., Choi, S.Y. and Han, J.S. (2006). DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiology Diseases, 22: 463-472.
- Ortiz-Abalia, J., Sahun, I., Altafaj, X., Andreu, N., Estivill, X., Dierssen, M. and Fillat, C., (2008). Targeting Dyrk1A with AAVshRNA attenuates motor alterations in TgDyrk1A, a mouse model of Down syndrome. American Journal Human Genetics, 83: 479-488.
- Guedj, F., Sébrié, C., Rivals, I., Ledru, A., Paly, E., Bizot, J.C., Smith, D., Rubin, E., Gillet, B., Arbones, M. and Delabar, J.M. (2009). Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PloS One, 4: e4606.
- Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R.C. and Garner, C.C., (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience, 10: 411-413.
- Rueda, N., Florez J. and Martinez-Cué, C. (2008). Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neuroscience Letters, 433: 22-27.
- Costa, A.C.S., Scott-McKean, J.J., and Stasko M.R. (2008). Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a conditioning fear test. Neuropsychopharmacology, 33: 1624-1632.
- Rueda, N., Llorens-Martín, M., Flórez, J., Valdizán, E., Banerjee, P. et al.(2010). Memantine normalizes several phenotypic features in the Ts65Dn mouse model of Down syndrome. J. Alzheimer Disease, 21(1): 277-290.
- Bianchi, P., Ciani, E., Guidi, S., Trazzi, S., Felice, D., Grossi, G., Fernandez, M., Giuliani, A., Calzà, L. andBartesaghi R. (2010). Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for Down syndrome. Journal of Neuroscience, 30(26): 8769-8779.
- Guidi, S., Stagni, F., Bianchi, P., Ciani, E., Giacomini, A., De Franceschi, M., Moldrich, R.,Kurniawan, N., Mardon, K., Giuliani, A., Calza, L., and Bartesaghi, R. (2014). Prenatal pharmacotherapy rescues brain development in a Down’s syndrome mouse model. Brain, 137(Pt 2): 380-401.
- Begenisic, T., Baroncelli, L., Sansevero, G., Milanese, M., Bonifacino, T., Bonanno, G., Cioni, G., Maffei, L. andSale, A. (2014). Fluoxetine in adulthood normalizes GABA release and rescues hippocampal synaptic plasticity and spatial memory in a mouse model of Down syndrome. Neurobiology Diseases, 63:12-19.



