
American Journal of Clinical Neurology and Neurosurgery, Vol. 1, No. 2, September 2015 Publish Date: Aug. 5, 2015 Pages: 81-85
Atypical Teratoid Rhabdoid Tumor with Atrial Septal Defect in a 6 Month Old Baby: A Surgical and Anesthetic Challenge
Ribhav Pasricha1, Guru Dutta Satyarthee1, *, Suvendu Purkait2, Mehar Chand Sharma2, Bhawani Shankar Sharma1
1Department of Neurosurgery, All India Institute of Medical Sciences, New Delhi, India
2Department of Neuropathology, All India Institute of Medical Sciences, New Delhi, India
Abstract
Atypical rhabdoid tumor is a malignant central nervous system tumor occurring predominantly in infants and carries high mortality and a poor prognosis. A 6-month old child who presented with posterior fossa mass lesion, was preoperatively detected to have coexistent patent foramen ovale and atrial septal defect. These congenital heart defects complicated the surgical and anesthetic management for the patient. The child underwent shunting for hydrocephalus followed by suboccipital craniectomy with gross total excision. The histopathology showed atypical teratoid/rhabdoid tumor, as confirmed by the loss of INI1 expression in the tumor cells. These tumors are highly malignant and carry very poor prognosis for the patients, most of whom are less than 2 years of age at diagnosis. The mean survival following surgery in these tumors is 11 months, although recent series suggest longer survival with trimodality therapy involving complete surgical resection; intravenous and intrathecal chemotherapy; and intensity-modulated radiotherapy.
Keywords
Infant, Atypical Rhabdoid Teratoma, Congenital Heart Disease, Patent Foramen Ovale, Atrial Septal Defect,
Atypical Teratoid Rhabdoid Tumor, ATRT
Received: July 15, 2015
Accepted: July 27, 2015
Published online: August 5, 2015
@ 2015 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY-NC license. http://creativecommons.org/licenses/by-nc/4.0/
1. Introduction
Atypical teratoid rhabdoid tumor (ATRT) is a malignant central nervous system tumor occurring predominantly in infants and accounts for 1-2% of all brain tumors, and at least 10% of central nervous system (CNS) tumors in infants, with a slight male predominance [10]. The overall prognosis of AT/RT is dismal, and the reported median survival is less than one year [4]. Distinguishing this tumor from radiologically similar lesions like medulloblastoma and CNS-PNET usually requires histopathology and is vital as ATRT carry a much worse prognosis. [1] We report the first known case in literature of a 6-month old child with a posterior fossa atypical rhabdoid tumor, who was preoperatively detected to have patent foramen ovale and atrial septal defect, and who underwent suboccipital craniectomy with gross total excision.
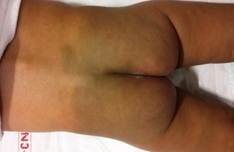
Figure 1. Clinical photograph showing the Mongolian spot over the child’s buttocks (congenital dermal melanocytosis).
2. Case Report
A 6-month male infant presented with refusal to feed, lethargy and repeated episodes of vomiting for three weeks. On examination the baby was drowsy and lethargic, but spontaneously moving all four limbs. His pupils were mid dilated and sluggishly reacting to light. His head circumference was 52 cm with tense and bulging anterior fontanelle. A large Mongolian spot was present on his buttocks (congenital dermal melanocytosis) [Fig 1]. NCCT head showed a large posterior fossa mass lesion arising from the vermis filling the fourth ventricle and growing superiorly and causing expansion of the aqueduct and reaching up to the third ventricle with associated obstructive hydrocephalus [Fig 2]. MRI brain showed a lesion in the superior vermis region, which was isointense on T1wI, causing expansion of the fourth ventricle and aqueduct of Sylvius [Fig 3]. On T2wI, multiple flow voids were observed [Fig 4a, 4b, 4c]. On contrast administration, the lesion showed no enhancement. There was restricted diffusion on DWI sequencing [Fig 5]. The child was planned for CSF diversionary procedure to relieve hydrocephalus followed by definitive surgery.
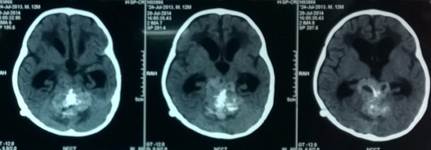
Figure 2. Radiograph of NCCT Head showing large posterior fossa tumor and obstructive hydrocephalus.
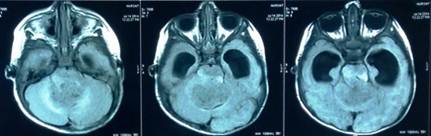
Figure 3. Axial T1w MRI image showing hypo- to isointense posterior fossa lesion compressing cerebellum, fourth ventricle and brainstem.
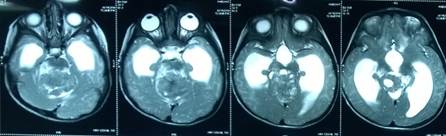
Fig 4a
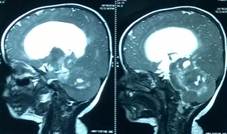
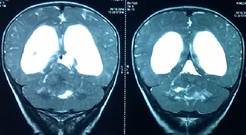
Fig 4b Fig 4c
Figure 4. Axial (Fig 4a), sagittal (Fig 4b), and coronal (Fig 4c) T2w MRI images showing the extent of the large tumor.
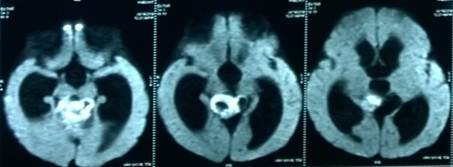
Figure 5. Diffusion weighted axial MRI image showing restricted diffusion suggesting highly cellular tumor.
During pre-anesthetic checkup, the child was found to have an ejection systolic murmur in the aortic area. An echocardiography was obtained which revealed a moderately sized ostium secundum atrial septal defect measuring 3.5 mm with left to right shunting of blood. There was also a patent foramen ovale.
A medium pressure ventriculoperitoneal shunt was inserted. Four days following shunt surgery, he was taken up for craniotomy and tumor resection. Due to the heart disease, the procedure could not be performed in sitting position to preclude the risk of air embolism taxing an already compromised heart. Adequate oxygenation had to be ensured to prevent hypoxia from setting in and this was done by the anesthetists ensuring good hydration and keeping the hemoglobin above 10 g/dl. Nitrous oxide was not used as anesthetic agent during surgery.
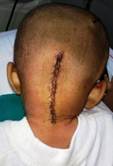
Figure 6. Postoperative image showing the skin incision for the midline suboccipital craniotomy.
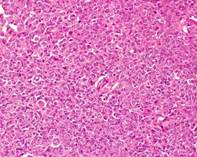
Figure 7a. Photomicrograph showing a highly cellular tumor with tumor cells arranged in sheets.
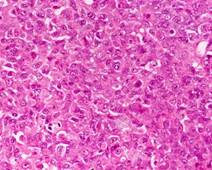
Figure 7b. Individual tumor cells showing eccentrically placed nuclei containing vesicular chromatin, prominent nucleoli and abundant cytoplasm (rhabdoid morphology). Few cells also showing eosinophilic globular cytoplasmic inclusion.
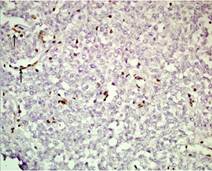
Figure 7c. The tumor cells showing loss of INI-1 expression. The INI-1 staining is retained in intratumoral endothelial cells (arrow).
A midline suboccipital craniotomy was done with removal of foramen magnum and c1 posterior arch [Fig 6]. Cistern magna was opened after inferomedial dural incision, and CSF was drained. Further dural opening was carried out. Vermis was exposed after the cerebellar hemispheres were retracted on both sides. Midline incision over the superior vermis revealed the vermian tumor. It was a bluish grey, soft, suckable, highly vascular tumor with good plane of dissection between the capsule and the surrounding normal tissue. Tumor was seen extending into the floor of the fourth ventricle and infiltrating into it at the lower pons level. Tumor was also infiltrating into the brainstem. A gross total resection was achieved, and surgery was completed uneventfully. He was electively ventilated overnight and extubated the next day. The patient’s sensorium and feeding improved a couple of days after surgery. However, on the third postoperative day, the patient developed transient cerebellar mutism which improved in the next two weeks. The child was discharged in a stable condition and started on adjuvant intravenous chemotherapy. The patient’s histopathology revealed an atypical rhabdoid/teratoid tumor, seen as a highly cellular tumor with tumor cells arranged in sheets with eccentric nuclei and abundant cytoplasm [Fig 7a, 7b, 7c]. Confirmatory was loss of INI1 expression in the tumor cells, as recent studies have demonstrated abnormalities of the INI1 gene on chromosome 22 in both CNS and non-CNS teratoid/rhabdoid tumors. [7]
3. Discussion
Atypical teratoid/rhabdoid tumors are rare highly aggressive neoplasms, accounting for 1-2% of all brain tumors, and about 10% of central nervous system (CNS) tumors in infants, even though tumors may sometimes present in older patients [1,11,12,17]. Their most common location is in the posterior fossa although some tumors have also been found in other locations like the supratentorial compartment, along cranial nerves, and in the spine [2,13,14,16]. The tumor was defined for the first time by Rorke et al [3]. These tumors are highly malignant and carry very poor prognosis for the patients, most of whom are less than 2 years of age diagnosis. The mean survival following surgery in these tumors is 11 months. [4] With trimodality therapy involving complete surgical resection; intravenous and intrathecal chemotherapy; and intensity-modulated radiotherapy, patients have shown longer disease-free survival in recent series [15,18,19,20].
These tumors may be associated with renal and extrarenal rhabdoid tumors which share the same genetics and imuunohistochemistry as these tumors [6]. There is also a case report of association with congenital unilateral cataract [5]. However, there has been no report in literature of these tumors being associated with congenital heart anomalies like atrial septal defects or patent foramen ovale. Our patient had both these findings along with congenital dermal melanocytosis which suggests a congenital origin for his cranial tumor as well.
Mongolian spots consist of blue-gray macular pigmentation. Mongolian spot is a hereditary developmental condition caused by entrapment of melanocytes in the dermis during their migration from the neural crest into the epidermis. The distinctive skin discoloration is due to the deep placement of the pigment in the dermis, which imparts a bluish tone to the skin. Typically, it is a few centimeters in diameter, although much larger lesions can also occur. Lesions may be solitary or numerous. Most commonly, Mongolian spots involve the lumbosacral area, but the buttocks, flanks, and shoulders may be affected in extensive lesions. Mongolian spots have been associated with cleft lip, spinal meningeal tumors and melanomas. A few cases of extensive Mongolian spots have been reported with inborn errors of metabolism, Hurler syndrome, gangliosidosis type 1, and Niemann-Pick disease [8, 9].
4. Conclusion
Atypical teratoid rhabdoid tumors are aggressive tumors found in infants with extremely poor long term outcomes. Surgery should aim at complete tumor resection; and with postoperative chemoradiation, a higher proportion of patients show long term survival. The presence of coexistent congenital heart disease complicates the operative management of these infants and appropriate anesthetic and surgical considerations need to be ensured for a good outcome.
References
- Al-Hussaini M, Dissi N, Al-Jumaily U, Swaidan M. Atypical teratoid rhabdoid tumor in childhood, 15 cases of a single institute experience.Turk Patoloji Derg. 2014;30(1):43-54.
- Yang M, Chen X, Wang N, Zhu K, et al. Primary atypical teratoid/rhabdoid tumor of central nervous system in children: a clinicopathological analysis and review of literature in China. Int J Clin Exp Pathol. 2014 Apr 15;7(5):2411-20. eCollection 2014.
- Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996 Jul;85(1):56-65.
- Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, Kun LE, Perlman EJ. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998 Sep;22(9):1083-92.
- Singh A, Jairajpuri Z, Gupta V, Sharma S, Chand K. Atypical teratoid/rhabdoid tumor of the central nervous system associated with congenital cataract. Indian J Pathol Microbiol. 2008 Jul-Sep;51(3):389-91.
- Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002 Nov;8(11):3461-7.
- Biegel, J.A., Zhou, J.Y., Rorke, L.B., Stenstrom, C., Wainwright, L.M., Fogelgren, B. (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 59:74–79.
- Igawa HH, Ohura T, Sugihara T, Ishikawa T, Kumakiri M. Cleft lip mongolian spot: mongolian spot associated with cleft lip. J Am Acad Dermatol. Apr 1994;30(4):566-9.
- Mosher DB, Fitzpatrick TB, Yoshiaki H, et al. Disorders of pigmentation. In: Fitzpatrick TB, ed. Dermatology in General Medicine. Vol 1. New York, NY: McGraw-Hill; 1993:903-95.
- Judkins AR, Eberhart CG, Wesseling P. Atypical teratoid /rhabdoid tumor. In Louis DN, Ohgaki H, Wiestler OD , Canavee WK, editors. WHO classification of tumours of the central nervous system. Lyon: IARC; 2007. 147-9.
- Udaka YT, Yoon JM, Malicki DM, Khanna PC, Levy ML, Crawford JR. Atypical Teratoid Rhabdoid Tumor in a Teenager with Unusual Infiltration Into the Jugular Foramen. World Neurosurg. 2015 Jul 14. pii: S1878-8750(15)00881-5.
- Afif M, Khalil J, Kouhen F, Aissa A, Omour Y et al. About a rare case of atypical rhabdoid teratoid tumor of the central nervous system in a pregnant woman. Pan Afr Med J. 2015 Jan 5;20:2.
- Oh CC, Orr BA, Bernardi B, Garré ML, Rossi A, Figà-Talamanca L, Robinson GW, Patay Z. Atypical teratoid/rhabdoid tumor (ATRT) arising from the 3rd cranial nerve in infants: a clinical-radiological entity? J Neurooncol. 2015 Jul 7.
- Dho YS, Kim SK, Cheon JE, Park SH, Wang KC, Lee JY, Phi JH. Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst. 2015 May 8.[Epub ahead of print].
- Verma V, Johnson CP, Bennion NR, Bhirud AR, Li S, McComb RD, Lin C. Atypical teratoid rhabdoid tumor: long-term survival after chemoradiotherapy. Childs Nerv Syst. 2015 May 5. [Epub ahead of print].
- Mahdi Y, Kharmoum J, Alouan A, Elouarradi H, Elkhiyat I et al. Primary atypical teratoid/rhabdoid tumor of the optic nerve: a rare entity in an exceptional location. Diagn Pathol. 2015 May 2;10:47.
- Jin S, Sun C, Yu S, Wang Q, An T, Wen Y. Atypical teratoid/rhabdoid tumor of the brain in an adult with 22q deletion but no absence of INI1 protein: a case report and review of the literature. Folia Neuropathol. 2015;53(1):80-5.
- Valencia-Moya A, González-García L, Ros-López B, Acha-García T, Weil-Lara B, Obando-Pacheco P, Arráez-Sánchez MÁ. Prognosis of atypical teratoid rhabdoid tumors (AT/RT) treated with multimodal therapy protocols. Report of our series. Neurocirugia (Astur). 2015 Apr 17. [Epub ahead of print].
- Lafay-Cousin L, Fay-McClymont T, Johnston D, Fryer C, Scheinemann K et al. Neurocognitive evaluation of long term survivors of atypical teratoid rhabdoid tumors (ATRT): The Canadian registry experience. Pediatr Blood Cancer. 2015 Jul;62(7):1265-9.
- Biswas A, Julka PK, Bakhshi S, Suri A, Rath GK. Intracranial atypical teratoid rhabdoid tumor: current management and a single institute experience of 15 patients from north India. Acta Neurochir (Wien). 2015 Apr;157(4):589-96.



