
International Journal of Preventive Medicine Research, Vol. 1, No. 2, June 2015 Publish Date: May 14, 2015 Pages: 17-21
Toxic Gangrenous Megacolon: A Novel Surgical Emergency in MELAS
Sivakumar Gopalswamy1, *, Joseph Mathew2
1Department of Surgery, Royal Cornwall Hospital, Truro, Cornwall, UK
2Department of Pathology, Royal Cornwall Hospital, Truro, Cornwall, UK
Abstract
MELAS syndrome (mitochondrial cytopathy, encephalomyopathy, lactic acidosis and stroke like episodes) is a syndrome in which signs and symptoms of gastro-intestinal disease are uncommon, if not rare. We describe a young female patient who presented as an acute surgical emergency, diagnosed as toxic mega colon necessitating an emergency total colectomy; the diagnosis of MELAS syndrome was made post-operatively. This diagnosis should be considered in patients presenting with acute ischemic colitis or toxic mega colon who have persistent lactic acidosis with encephalopathy and a stormy post-operative course.
Keywords
Encephalopathy, Lactic Acidosis, MELAS, Toxic Colon
Received: April 3, 2015 / Accepted: April 19, 2015 / Published online: May 11, 2015
@ 2015 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY-NC license. http://creativecommons.org/licenses/by-nc/4.0/
1. Introduction
MELAS syndrome (mitochondrial cytopathy, encephalomyopathy, lactic acidosis and stroke like episodes) is principally a progressive, neurodegenerative disease in which gastro-intestinal involvement is not a major feature but in which vomiting, anorexia and pseudo-obstruction and colitis have been reported 1. We describe the case of a young female who presented with an acute surgical abdomen, in whom a diagnosis of acute colitis or toxic mega colon made on clinical and imaging grounds. Total colectomy performed and histology confirmed ischaemic colitis. Persistent lactic acidosis and encephalopathy prompted further neurological evaluation. Subsequent intestinal, muscle biopsy, and genetic studies confirmed the diagnosis of MELAS syndrome. This report highlights the uncommon presentation, diagnostic difficulties and significant morbidity involved with colitis in MELAS syndrome.
2. Case Presentation
A 34-year-old Caucasian woman presented with sudden onset of lower abdominal pain and vomiting of 12 hrs duration. Clinical examination revealed diffuse tenderness with rebound signs in the lower abdomen. She went on to have an episode of collapse from which resuscitated and taken to intensive care, sedated, intubated and ventilated.
Initial haematology and biochemical results were Hb: 19.6 g/dl, WCC: 42.1x109, /L, amylase 110 IU/L, CRP 15mg/L and corrected calcium 3.67. CT abdomen revealed a markedly dilated colon and rectum with thickening of its wall and enhancement suggestive of toxic colitis. Normal enhancement of the major mesenteric arteries noted. In intensive care, she was severely acidotic with blood gas analysis showing a pH 7.09, lactate levels of 15 mmol/L and base excess of 22. This started to normalise with large amounts of fluid. She maintained a good urine output and her tachycardia and blood pressure normalised. She was in distress and developed diarrhoea after admission. Subsequent flexible sigmoidoscopy showed ischemic mucosa from the recto sigmoid junction to the descending colon. A diagnosis of acute colitis or toxic mega colon secondary to ulcerative colitis made.
She had several episodes of abdominal pain over the last few weeks and treated by her practitioner as irritable bowel. She had a normal childhood developmental history and performed well in school. She was on Fluoxetine 20 mg daily for depression over the last 10 years. Four years previously, a brain MRI performed for persistent galactorrhoea, which ruled out a pituitary mass but showed basal ganglia calcification; she was also diagnosed to have bilateral neurosensory loss.
The following day she underwent a laparotomy and found to have an ischaemic colon; a total colectomy and end ileostomy performed. The rectal stump stapled and anchored in the subcutaneous area. Histological examination showed extensive ischaemic change in the colonic wall with minimal inflammation and normal blood vessels (figure 1). The postoperative period was characterised by persistent lactic acidosis, decreased level of consciousness, despite withdrawal of sedation and needed ventilator support for 12 days.
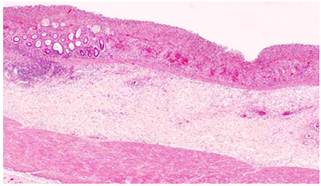
Figure 1. Extensive ischemic change in the colonic wall with minimal inflammation and normal blood vessels.
The triad of ischaemic colitis, lactic acidosis and abnormal brain scan prompted a neurology review; the possibility of a mitochondrial disorder considered based on the postoperative events, previous deafness and calcification of basal ganglia. She subsequently needed a tracheostomy and developed adult respiratory distress syndrome. CT brain revealed extensive calcification of basal ganglia, periventricular calcification and calcification extending into cerebellum, diffuse involutional changes noted in the ventricles and sulci. A subsequent skeletal muscle biopsy revealed ragged red fibres (figure 2), COX -negative fibres, SDH hyper reactive blood vessels and Para crystalline arrays within mitochondria.Genetic studies revealed an A3243G mutation characteristic of MELAS.
She stayed in ITU for 55 days and sent to a rehabilitation ward for intensive physiotherapy. She presented seven months later with sudden onset of central abdominal pain and vomiting and managed conservatively as adhesive partial bowel obstruction. Currently she is requesting a reversal of ileostomy.
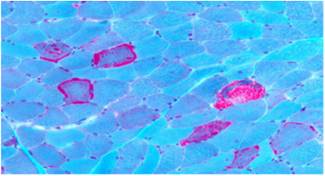
Figure 2. Gomori trichrome preparation showing scattered ‘ragged red’ fibres.
3. Discussion
Mitochondrial cytopathies are a diverse group of inherited and acquired disorders of the mitochondria, which are characterised by protean clinical manifestations and inadequate energy production 1. The diagnostic criteria for MELAS syndrome include 2:
1). Stroke like episodes,
2). Encepahalopathy with seizures and dementia,
3). Mitochondrial myopathy characterised by lactic acidosis, ragged red fibres on muscle biopsy, accumulation of abnormal mitochondria in smooth muscles and endothelial cells and, mutations of mitochondrial DNA detected at genetic testing.
Multisystemic organ involvement is seen, including the CNS, skeletal muscle, eye, cardiac muscle, and, more rarely, the GI and renal systems.3, 4Sensorineural hearing loss, non-insulin dependent diabetes mellitus, urinary retention, recurrent headaches, vomiting and migraine are other associated features of MELAS syndrome 1,5.MELAS presentation occurs with the first stroke like episode, usually when an individual is aged 4-15 years. Less often, onset of disease may occur in infancy with delayed developmental milestones and learning disability. Short stature may be the first manifestation of MELAS syndrome in many patients. Patients may develop features of hypothyroidism and hyperthyroidism.
MELAS syndrome has been associated with at least 6 different point mutations, 4 of which are located in the same gene, the tRNALeu (UUR) gene. Approximately 80% of patients with the clinical characteristics of MELAS syndrome have a heteroplasmic A-to-G point mutation in the dihydrouridine loop of the transfer RNA (tRNA)Leu (UUR)gene at base pair (bp) 3243 (ie, 3243 A → G mutation).6However, other mitochondrial DNA (mtDNA) mutations are observed, including the m.3244 G → A, m.3258 T → C, m.3271 T → C, and m.3291 T → C in the mitochondrial tRNALeu(UUR)gene.7 In Northern England, the prevalence of 3243A → G mutation in the adult population has been determined to be approximately 1 per 13,000.
Mutations in MELAS affect mitochondrial tRNA function, leading to the disruption of the global process of intramitochondrial protein synthesis. Measurements of respiratory enzyme activities in intact mitochondria have revealed that more than one half of the patients with MELAS syndrome may have complex I or complex I + IV deficiency. A close relationship is apparent between MELAS and complex I deficiency. The decreased protein synthesis may ultimately lead to the observed decrease in respiratory chain activity by reduced translation of UUG-rich genes such as ND6 (component of complex I).8
The pathogenesis of the stroke like episodes in MELAS syndrome has not been completely elucidated.9 The unusual stroke like episodes and higher morbidity observed in MELAS syndrome may be secondary to alterations in nitric oxide homeostasis that cause microvascular damage. Nitric oxide can bind the cytochrome c oxidase–positive sites in the blood vessels present in the CNS, displacing heme-bound oxygen and resulting in decreased oxygen availability in the surrounding tissue and decreased free nitric oxide. Also, coupling of the vascular mitochondrial dysfunction with cortical spreading depression might underlie the selective distribution of ischemic lesions in the posterior cortex in these subjects.
Other abnormalities that may be observed are ventricular dilatation, cortical atrophy, and basal ganglia calcification. Mental deterioration usually progresses after repeated episodic attacks. Psychiatric abnormalities and cognitive decline (eg, altered mental status, schizophrenia) may accompany the stroke like episodes. Bipolar disorder is another psychiatric abnormality observed in MELAS syndrome. Autism spectrum disorders (ASDs) with or without additional neurological features can be early presentations of the m.3243 A → G mutation. Myopathy may be debilitating. The encephalopathy may progress to dementia; eventually, the clinical course rapidly declines, leading to severe disability and premature death. The progressive disorder has a high morbidity and mortality. The encephalomyopathy, associated with stroke like episodes followed by hemiplegia and hemianopia, is severe. Focal and general convulsions may occur in association with these episodes. Skin manifestations of cutaneous purpura, hirsutism, and a scaly, pruritic, diffuse erythema with reticular pigmentation may be observed in patients with MELAS syndrome.
Another cause of high mortality is the less common feature of cardiac involvement, which can include hypertrophic cardiomyopathy, hypertension, and conduction abnormalities, such as atrioventricular blocks, long QT syndrome, or Wolff-Parkinson-White syndrome. Subjects with MELAS syndrome were found to have increased ascending aortic stiffness and enlarged aortic dimensions suggesting vascular remodeling. Aortic root dissection was found in one patient with MELAS syndrome.10Some patients may develop Leigh syndrome (ie, subacute necrotizing encephalopathy). Patients may develop renal failure due to focal segmental glomerulosclerosis.
More rarely, these patients may exhibit severe GI dysmotility and endocrine dysfunction. Mitochondrial disorders associated with gastro-intestinal involvement 1,5,11,12 including recurrent vomiting, anorexia, peptic ulceration, severe constipation, paralytic ileus and colonic volvulus. MNGIE (mitochondrial neurogastrointestinal encephalopathy)2,11, an autosomal recessive mitochondrial disorder typically has visceral neuropathy characterised by diarrhoea, vomiting, constipation, intermittent intestinal obstruction, gastro paresis, ileus and colitis.
Gastro-intestinal involvement albeit rare, can be the initial presenting feature in MELAS syndrome 1,12. Surgical intervention in MELAS syndrome has been required occasionally in patients with ischaemic colitis, segmental ileal paralysis, pseudo-obstruction, gastric perforation and mega colon 1,5. The presence of mutated mitochondrial DNA leading on to smooth muscle dysfunction is the likely mechanism for paralytic ileus 13. Autonomic neuropathy 2, focal accumulation of mitochondrial A3243G mutations in stomach 14,15 and colon 1 are also reported to be a contributory factor in gastric dysmotility and ischaemic colitis in MELAS syndrome.
The following studies are indicated in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) syndrome:
span style='font:7.0pt "Times New Roman"'> In muscle biopsies stained with hematoxylin and eosin, variation is observed in type 1 and type 2 fiber sizes, representing myopathic changes.
span style='font:7.0pt "Times New Roman"'> Ragged red fibers are the hallmark of MELAS syndrome. The ragged red fibers stain brilliant red with occasional cytoplasmic bodies with trichrome stain. Ragged red fibers usually stain positive with cytochrome oxydase stain.
span style='font:7.0pt "Times New Roman"'> Serum lactic acid, serum pyruvic acid, cerebrospinal fluid (CSF) lactic acid, and CSF pyruvic acid
span style='font:7.0pt "Times New Roman"'> The levels of serum creatinekinase are mildly to moderately increased in some patients with MELAS syndrome
span style='font:7.0pt "Times New Roman"'> Individuals with more severe clinical manifestations of MELAS syndrome generally have greater than 80% mutant mtDNA in stable tissues such as muscle.
span style='font:7.0pt "Times New Roman"'> If a muscle biopsy is performed to pursue a diagnostic evaluation, then test respiratory chain enzyme activities.
The following imaging studies may be indicated:
span style='font:7.0pt "Times New Roman"'> CT scan or MRI of the brain following a stroke like episode reveals a lucency that is consistent with infarction. Patients with MELAS syndrome who have a presentation similar to Leigh syndrome may have calcifications in the basal ganglia.
span style='font:7.0pt "Times New Roman"'> PET studies may reveal a reduced cerebral metabolic rate for oxygen.
span style='font:7.0pt "Times New Roman"'> Single-photon emission computed tomography (SPECT) studies can ascertain strokes in individuals with MELAS syndrome using a tracer, N -isopropyl-p-[123-I]-iodoamphetamine.
span style='font:7.0pt "Times New Roman"'> Proton magnetic resonance spectroscopy (1 H-MRS): This is used to identify metabolic abnormalities, including the lactate-to-creatine ratio in either muscle or brain and the decreased CNS N -acetylaspartate–to–creatine ratio in regions of stroke. With this technique, elevated regions of lactate have been detected while serum levels are normal.
span style='font:7.0pt "Times New Roman"'> Echocardiography: This is useful to evaluate for hypertrophic and dilated cardiomyopathy and aortic root dimensions; however, cardiomyopathy is not a common feature in individuals with MELAS syndrome. ECG is used to look for conduction abnormalities with ventricular arrhythmias. ECG can identify presymptomatic cardiac involvement, preexcitation syndromes, and cardiac conduction block.
span style='font:7.0pt "Times New Roman"'> EEG findings are usually abnormal. Epileptiform spike discharges are usually present.
For individuals with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS) syndrome and for those with other oxidative phosphorylation (OXPHOS) disorders, metabolic therapies are administered to increase the production of adenosine triphosphate (ATP) and to slow or arrest the deterioration of this condition and other mitochondrial encephalomyopathies. Metabolic therapies used for the management of MELAS syndrome include carnitine, CoQ10, phylloquinone, menadione, ascorbate (ie, ascorbic acid), riboflavin, nicotinamide, creatine monohydrate, idebenone, succinate, and dichloroacetate. However, assessment of the efficacy of these compounds is far from complete, and efficacy is believed to be limited to individual cases.
Treatment with CoQ10 has been helpful in some patients with MELAS syndrome. No adverse effects have been reported from its administration. Menadione (vitamin K-3), phylloquinone (vitamin K-1), and ascorbate have been used to donate electrons to cytochrome c. Idebenone has also been used to treat this condition, and improvements in clinical and metabolic abnormalities have been reported. Riboflavin has been reported to improve the function of a patient with complex I deficiency and the m.3250 T → C mutation. Nicotinamide has been used because complex I accepts electrons from nicotinamide adenine dinucleotide (NADH) and ultimately transfers electrons to Q10. Dichloroacetate is another compound used with these agents, because levels of lactate are lowered in plasma and cerebrospinal fluid (CSF). Patients reportedly may respond in a favorable manner. Patients with MELAS may have less chance of having stroke like episodes by improving their endothelial function with oral supplementation of L-arginine.
4. Conclusion
Clinicians managing patients with either acute or chronic gastro-intestinal symptoms not attributable to a specific cause should maintain a high index of suspicion of mitochondrial cytopathy 12. Recurrent vomiting, diarrhoea, pseudo-obstruction, or an acute abdomen associated with lactic acidosis should prompt a neurological assessment with histological and genetic evaluation to rule out the possibility of MELAS syndrome. MELAS syndrome widely varies in presentation. Patients in general tend to have a poor prognosis and outcome. The encephalomyopathy tends to be severe and progressive to dementia. Currently, no therapies have proven efficacy. The prognosis especially in patients with sepsis and metabolic disturbances undergoing surgery is unfavorable 16; encephalomyopathy and cardiac involvement in MELAS is associated with a high morbidity and mortality 2.
Acknowledgements
Professor Dough Turbull, Neurologist, Mitochondrial disease research group, University of Newcastle upon Tyne, United Kingdom.
References
- Hess J, Burkhard P, Morris M, Lalioti M, Myers P, Hadengue A. Ischaemic colitis due to mitochondrial cytopathy. Lancet 1995; 346:189-90.
- Cohen BH, Gold DR. Mitochondrial cytopathy in adults: what we know so far. Cleve Clin J Med 2001; 68:625-6.
- Seidowsky A, Hoffmann M, Glowacki F, Dhaenens CM, Devaux JP, Lessore de Sainte Foy C, et al. Renal involvement in MELAS syndrome - a series of 5 cases and review of the literature. Clin Nephrol. Aug 21 2012.
- Meseguer S, Martínez-Zamora A, García-Arumí E, Andreu AL, Armengod ME. The ROS-sensitive microRNA-9/9* controls the expression of mitochondrial tRNA-modifying enzymes and is involved in the molecular mechanism of MELAS syndrome. Hum Mol Genet. Aug 22 2014.
- Garcia-Velasco A, Gomez-Escalonilla C, Guerra-Vales JM, Cabello A, Campos Y, Arenas J.Intestinal pseudo-obstruction and urinary retention: cardinal features of a mitochondrial DNA-related disease. J Intern Med 2003 Mar; 253:381-5.
- Mehrazin M, Shanske S, Kaufmann P, Wei Y, Coku J, Engelstad K. Longitudinal changes of mtDNA A3243G mutation load and level of functioning in MELAS. Am J Med Genet A. Feb 15 2009;149A(4):584-7.
- Liu K, Zhao H, Ji K, Yan C. MERRF/MELAS overlap syndrome due to the m.3291T>C mutation. Metab Brain Dis. Mar 2014;29(1):139-44.
- Sasarman F, Antonicka H, Shoubridge EA. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum Mol Genet. Dec 1 2008;17(23):3697-707.
- Testai FD, Gorelick PB. Inherited metabolic disorders and stroke part 1: Fabry disease and mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Arch Neurol. Jan 2010;67(1):19-24.
- Nemes A, Geleijnse ML, Sluiter W, Vydt TC, Soliman OI, van Dalen BM. Aortic distensibility alterations in adults with m.3243A>G MELAS gene mutation. Swiss Med Wkly. Feb 21 2009;139(7-8):117-20.
- Chang TM, Chi CS, Tsai CR, Lee HF, Li MC. Paralytic ileus in MELAS with phenotypic features of MNGIE. Pediatr Neurol 2004; 31:374-7.
- Muehlenberg K, Fiedler A, Schaumann I, Muller-Felber W, Wiedmann KH. Intestinal pseudo-obstructions and gastric necrosis in mitochondrial myopathy Dtsch Med Wochenschr 2002 Mar 22; 127:611-5.
- Hiel JA, Verrips A, Keyser A, Jansen TL, Wesseling P, de Coo R, Gabreels FJ.Ileus in mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes. Neth J Med 1998; 53:27-31.
- Inoue K, Ikegami H, Fujisawa T, Kawabata Y, Shintani M, Nijima K, Masaya O, Nishino M, Itoi-Babaya M, Babaya N, Ogihara T. High degree of mitochondrial 3243 mutation in gastric biopsy specimen in a patient with MELAS and diabetes complicated by marked gastrointestinal abnormalities. Diabetes Care 2003; 26:2219.
- Fujii A, Yoneda M, Ohtani M, Nakagawa H, Kumano T, Hayashi K, Muramatsu A, Takabatake S, Ibi T, Sahashi K, Azuma T, Kuriyama M. Gastric dysmotility associated with accumulation of mitochondrial A3243G mutation in the stomach. Intern Med 2004; 43:1126-30.
- Shimotake T, Furukawa T, Inoue K, Iwai N, Takeuchi Y. Familial occurrence of intestinal obstruction in children with the syndrome of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). J Pediatr Surg 1998; 33:1837-9.



