
International Journal of Advanced Materials Research, Vol. 1, No. 3, July 2015 Publish Date: May 28, 2015 Pages: 73-79
One Step PCR for Detection of Staphylococcus aureus Specific Sequence Gene and MecA Gene
Kumurya A. S.1, *, Gwarzo M. Y.1, Uba A.2
1Department of Medical Laboratory Science, Faculty of Allied Health Sciences, Bayero University, Kano, Nigeria
2Biological Science Programme, School of Science, Abubakar Tafawa Balewa University, Bauchi, Nigeria
Abstract
Background: Methicillin – resistant Staphylococcus aureus (MRSA) has also been noted as one of the main pathogen of public health importance. Detection of the mecA gene by polymerase chain reaction (PCR) is the gold standard for identifying methicillin-resistant Staphylococcus aureus (MRSA). Objectives: In order to accelerate the procedure of identification in clinical microbiology laboratories, it is very important to havea simple and rapid method for DNA extraction. In this work, a one step PCR assay for the detection of clinically relevant antibiotic resistance genes (mecA gene) harboredby some Staphylococcus aureus isolates and for the simultaneous identification of such isolates at the species level has been described. Methods: In this study, a rapid method for bacterial DNA extraction directly from a single colony that gave quality DNA for PCR in as little as 0 minute was used. Polymerase chain reaction (PCR) was used to amplify both the S. aureus specific sequence gene and mecA gene of 100 isolates with the amplicon size of 107 and 532bp. Results: All the isolates (n=100) expressed S. aureus specific sequence gene in their PCR products. Only 5 isolates (5.0%) were confirmed as MRSA based on the detection of mecA gene.This protocol yielded good-quality target DNA for PCR amplification. Amplifications using that DNA gave rise to goodquantities of the expected PCR fragments. When PCR was performed using DNA obtained by this method or previously reported methods, no differences were observed. Conclusion: Nowadays, with only a few antibiotics such as vancomycin constituting the last defense against MRSA, and due to the increasing incidence and spread of MRSA, it is absolutely necessary that fast and sensitive laboratory methods are available for the immediate detection of multiple-antibiotic-resistant MRSA. This method reliably amplified DNA from Staphylococcus aureus colonies without a DNA extraction step. As our results showed, the method herein described is highly sensitive, specific, fast, and feasible. Hence, considering that it represents a rapid, simple, and cost-effective method, it could be systematically applied in clinical microbiology laboratories for the identification of MRSA, bringing insights into antibiotic therapy design and helping treatment to be initiated without delay.
Keywords
Staphylococcus aureus, MRSA, MecA Gene, DNA, PCR
Received: April 9, 2015
Accepted: April 24, 2015
Published online: May 27, 2015
@ 2015 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY-NC license. http://creativecommons.org/licenses/by-nc/4.0/
Contents
1. Introduction 2. Materials and Methods 2.1. The Study of Areas 2.2. Specimen Collection 2.3. Identification of Staphylococcus aureus Isolates and Susceptibility Testing 2.4. Storage of Staphylococcus aureus Isolates 2.5. Rapid DNA Extraction Method 2.6. Oligonucleotide Primers 2.7. PCR Amplification 3. Results 4. Discussion 5. Conclusion Acknowledgments
1. Introduction
Methicillin – resistant Staphylococcus aureus (MRSA) has also been noted as one of the main pathogen of public health importance. It was first discovered in the United Kingdom in 1961 and is now widespread, particularly in the hospital setting where it is commonly termed a super bug (Jerons, 1991). The term methicillin resistant is historically used to describe resistance to any of this class of antimicrobials. Today in USA approx 35% of hospital strains of S. aureus are resistant to methicillin (or other penicillin antibiotics). And in recent years the emergence of vancomycin resistant S. aureus(VRSA) has caused additional concern. Non methicillin – resistant strains of Staphylococcus aureus are called methicillin susceptible Staphylococcus aureus (MSSA). Patients with MRSA infections have been noted with worse clinical and economical outcome compared with patients with MSSA infections. A study by Chang et al. (2004) indicated that bacterial meningitis caused by MRSA was associated with a mortality rate of 56% compared to a mortality rate of 135 in a patient group with meningitis cause by MSSA. Also some Cohort studies of patients with MRSA bacteria have reported higher mortality rates increased morbidity. Longer hospital stay and higher cost compared with patients with MSSA bacteria (Blot et al., 2002; Melzer et al., 2003; Kopp et al., 2004). In addition, MRSA infections are particularly difficult to treat if they are at anatomical sites, where antibiotic penetration is reduced (Duckworth, 2003).Detection of the mecA gene by PCR has been described as a rapid method for the identification of MRSA(Tokue et al., 1991; Brakstad et al., 1992; Tokue et al., 1992;Unalet al., 1994;Barski et al., 1996; York et al., 1996; Kearns et al., 1999; Martineau et al., 1998; Kohner et al., 1999).
Here, we describe a rapid and simple PCR assay for simultaneous detection of the S. aureus specific sequence gene and mecA using a method of extracting DNA directly from a singlecolony.
2. Materials and Methods
2.1. The Study of Areas
A total of 8 health institutions located in six states in Northwestern Nigeria, were enrolled in the study. The hospitals were two teaching hospitals, three Federal Medical Centers, two Specialist Hospitals and one Infectious Diseases Hospital.
2.2. Specimen Collection
A total of 100 consecutive non – duplicated Staphylococcus aureus isolates were obtained from clinical samples in the health institutions across Northwestern Nigeria. The isolates were collected during two years period from February 2008 to April 2010. Routine clinical microbiology specimens were selected during the period. The quality control and rejection criteria of specimen were followed (Isenberg, 1998). Staphylococcus aureus ATCC 25923 was used as a reference control organism. Specimens were processed within 2 hours of collection by the standard microbiology technique. The sheep blood agar and mannitol salt agar were used for inoculation and the agar plates were then incubated at 350C for 18-24 hours in aerobic atmosphere (NCCLS, 2003).
2.3. Identification of Staphylococcus aureus Isolates and Susceptibility Testing
Screening for methicillin resistance was done by 1µg oxacillin disk diffusion testing, in which the disk was placed on Mueller-Hinton agar (Difco Laboratories, Detroit, Mich.) and incubated for 24h at 30°C following the NCCLS guidelines(NCCLS,2002).Biochemical identifications of staphylococci were performed according to standard laboratory criteria (Cheesbrough, 2000). Staphylococcus aureus ATCC 25923 was used as a reference control organism.
2.4. Storage of Staphylococcus aureus Isolates
All confirmed Staphylococcus aureus isolates were stored in 16% v/v glycerol broth at -80oC in screw-cap bottles. The pH of the medium ranged from 7.2-7.6 at room temperature. Using sterile swab, the entire growth of an overnight pure culture was sub-cultured in 5ml of sterile glycerol broth and immediately stored in freezer [Micro bank (Diagnostic pro - lab)] at -80oC. After 24 hours the viability of the organism was checked by thawing the suspension at 35oC and inoculated on blood agar plates.
2.5. Rapid DNA Extraction Method
After overnight culture on brain heart infusion (Difco Laboratories) agar plates, one colony of each sample was directly used as a template for PCR amplification. DNA obtained by this method was compared with DNA obtained by previously reported methods (Bignardi et al., 1996; Perez et al., 2001; Anna-Kaarina et al., 2009).
2.6. Oligonucleotide Primers
The oligonucleotide primers used in this study have been previously described (Martineau et al., 1998; Meshref et al., 2011) and were obtained from acommercial source (Inqaba Biotechnical Industries (Pty) Ltd., South Africa). The 3-end region of the S. aureus specific gene was amplified using A30nucleotide forward primer 5’- AATCTTTGTCGGTACACG ATATTCTTCACG -3’ and A30 nucleotide reverse primer, 5’-CGTAAT GAG ATT TCAGTA GAT AATACAACA-3’ (which hybridize to 5-34 and (112-83), respectively, (Martineau et al., 1998). While The 3-end region of the mecAgene was amplified using A22nucleotide forward primer 5’- AAA ATC GAT GGT AAA GGTTGG C - 3’ and A22 nucleotide reverse primer, 5’- AGTTCTGCAGTACCG GAT TTG C-3’ (which hybridize to sites 1282-1301 and 1814-1793) (Robert Koch institute, 2003) (Table 1).Staphylococcus aureus specific gene and mecA gene have the amplicon size of 107 and 532bp using primers described by (Meshref et al., 2011).
Table 1. Oligonucleotide Primers used in the PCR assay
| Oligonucleotide | Sequence | Target |
| position | Nucleotide | Gene |
| A30fwd5-34 | AATCTTTGTCGGTACACGATATTCTTCACG | Sa |
| A30 rev112-83 | CGTAATGAGATTTCAGTAGATAATACAACA | Sa |
| A22fwd1282-1301 | AAAATCGATGGTAAAGGTTGGC | mecA |
| A22 rev1814-1793 | AGTTCTGCAGTACCGGATTTGC | mecA |
2.7. PCR Amplification
PCR assays were all directly performed from the bacterial suspension obtained after the rapid DNA extraction method described above. The DNA template was added to 100µlof PCR mixture consisting of 1× reaction buffer [16 mM (NH4)2SO4,67 mMTris-HCl (pH 8.8)], a 0.5mM concentration of each of the four deoxyribonucleoside triphosphates (dATP, dCTP, dGTP, and dTTP) (Inqaba Biotechnical Industries (Pty) Ltd., South Africa), 1.0μM of eachprimer, and mecA primer, and 1.25U of the Dream Taq™ Green PCR Master Mix (2x) (Fermentas Life Sciences, supplied by Inqaba Biotechnical Industries (Pty) Ltd., South Africa) is a ready-to-use solution containing Dream Taq™ DNA polymerase, optimized Dream Taq™ Green buffer and 4mMMgCl2. For each sample, one reactionwas performed with the pair of primers to identify S. aureusspecific sequence gene and with the mecA pairs of primers to detectthe resistance gene (mecA). In order to reduce the formation of nonspecificextension products, a hot-start PCR protocol was used; the tubeswere placed in the thermal cycler when the denaturing temperaturewas reached. All PCR assays were carried out with a negativecontrol containing all of the reagents without DNA template. DNAamplification was carried out in a Techne PCR system TC-5000 thermo cycler (Bibby Scientific Ltd.) with the following thermal cycling profile: an initial denaturation step at 94°C for 5 min (which lysed the bacterial cell) was followed by1 cycle of amplification this was followed by denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 60 s ending with a final extension step at 72°C for 5 min. After PCR amplification, 5µl was removed and subjected to agarose gel electrophoresis(1.5% agarose, 1× Tris-borate-EDTA, 100 V, 40 min) to estimatethe sizes of the amplification products by comparison with a 100-bp O’ GeneRuler™ 100bpmolecular size standard DNA Ladder, ready-to-use designed by Fermentas Life sciences (supplied by Inqaba Biotechnical Industries (Pty) Ltd., South Africa). The gel was stained with ethidium bromide, and the amp icons were visualized using a UV lightbox. This protocol, including the rapid DNA extraction method from a single colony and electrophoretic analysis of the amplified products on an agarose gel, was performed in less than 4hours.
3. Results
In this report, a rapid method for bacterial DNA extraction directly from a single colony that gave quality DNA for PCR in as little as 0 min was described. This protocol yielded good-quality target DNA for PCR amplification. Amplifications using that DNA gave rise to good quantities of the expected PCR fragments. When PCR was performed using DNA obtained by this method or previously reported methods, no differences were observed. The chromosomal DNA extraction from Staphylococcus aureus clinical isolates showed that all the isolates (n = 100) expressed S. aureus specific sequence gene of 107 base pairs (bp) in their PCR products, which confirmed the assumption that all the strains were Staphylococcus aureus. Fig. I shows a representative agarose gel electrophoresis of PCR products.
The chromosomal DNA extraction from Staphylococcus aureus clinical isolates showed different methicillin sensitivity results as compared to the disk diffusion method of MRSA detection used in the study. The DNA of the same 100 isolates was used as templates for PCR amplification with mecA primer pair. The size of the amplicon was expected to be 532 base pairs (bp). An amplicon of 532bp was seen in only 5 of the 100 isolates tested. PCR amplification of mecA gene demonstrating amplicon of 532bp products are given in Fig. 2
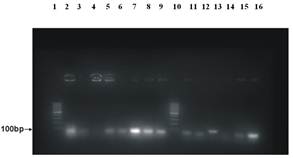
Figure 1. Agarose gel electrophoresis patterns showing PCR amplification products from S. aureus isolates. A 100 molecular weight size marker (O’ GeneRuler™ 100bp ladder) was applied at the lane 1 and 10 of the gel to identify the isolated genes. A negative control (methicillin susceptible S. aureus, ATCC 25923) was applied at the lane 13. PCR products of the test samples were applied on lane 2,3,4,5,6,7,8,9,11,12,14,15 and 16 which showed clear bands confirmed that all the isolates were Staphylococcus aureus.

Figure 2. Representative agarose gel electrophoresis of PCR products. Lanes 12, 14 are positive for mecA as indicated by 532bp PCR product (Methicillin Resistant S. aureus isolates), Lane 2-9, 11, 13, 15, are negative for mecA (Methicillin Susceptible S. aureus isolates), Lane 16: negative control (methicillin susceptible S. aureus, ATCC 25923); Lane 1, 10: molecular weight size marker.
The result of oxacillin and methicillin susceptibility tests (determined by the disk diffusion method for 100 staphylococcal isolates) was compared with the results obtained by the PCR assays for the detection of mecA gene. All the 100 staphylococcal isolates were resistant to oxacillin or methicillin using phenotypic assay. The isolates were later tested to confirm using PCR protocol. The isolates of Staphylococcus aureus that carry the mecA gene were reported as methicillin resistant while the isolates that did not carry mecA gene were reported as methicillin susceptible. Only 5 of the 100 Staphylococcus aureus isolates were found to have the PCR amplification of mecA gene demonstrating the expected 532bp product. For the strains included in this study, microbiological susceptibility testing and PCR results did not show concordant results (Table2). Therefore the prevalence rate of MRSA in current study was 5.0 %. Among the MRSA strains, three MRSA isolate was detected in wound samples while the remaining 2 MRSA isolates were detected in urine samples. The distribution of mecA-positive Staphylococcus aureus according to type of specimen is shown in Table 3. The statistical analysis of the result shows significant difference (p< 0.05) between mecA positive Staphylococcus aureus and mecA negative Staphylococcus aureus isolates.
Table 2. Correlation between phenotypic groups and PCR results
| No. of strains with result indicated | |||||
| MecA PCR result | No. of isolates tested | Methicillin Disc Diffusion was: | Oxacillin Disc Diffusion was: | ||
| Pos | Neg | Pos | Neg | ||
| Positive | 5 | 5 | 0 | 5 | 0 |
| Negative | 95 | 95 | 0 | 95 | 0 |
Key: Pos, Positive; Neg, Negative
Table 3. Distribution of Staphylococcus aureus isolates according to the type of specimen.
| Specimen type | Staphylococcus aureus isolates | ||
| No. of mecA+(%) | No. of mecA-(%) | Total(%) | |
| Wound swabs | 3 (9.7) | 28 (90.3) | 31 (31.0) |
| Ear swabs | 0 (0.0) | 9 (100) | 9 (9.0) |
| Blood culture | 0 (0.0) | 8 (100) | 8 (8.0) |
| Urine | 2 (7.7) | 24 (92.3) | 26 (26.0) |
| High vaginal swabs | 0 (0.0) | 12 (100) | 12 (12.0) |
| Sputum | 0 (0.0) | 11 (100) | 11 (11.0) |
| Semen | 0 (0.0) | 1 (100) | 1 (100) |
| Urethral swabs | 0 (0.0) | 2 (100) | 2 (100) |
| Total | 5 (5.0) | 95 (95.0) | 100 (100) |
Mean = 0.285714, SE = 0.285714, SD = 0.755929, CL (95.0%) =0.699118
Key:
mecA+=mecA positive
mecA-=mecANegative
4. Discussion
During the last decade, many studies have demonstrated the extremely high capacity of PCR for specifically detecting bacteria and genes of interest (Salisbury et al., 1996). That ability has revealed PCR asa powerful tool in clinical microbiology studies (Cockerill, 1999). Several authors have already shown the feasibility of the PCR methodology for the identification of S. aureus strains and for the detection of antibiotic resistance genes (Vannuffel et al., 1998; Cockerill, 1999;Jonas et al., 1999).PCR identification of S. aureus has been based on the detection of different specific target sequences such as nuc (Brakstad et al., 1992) and coaA (Tang et al., 1997; Tenover et al., 1999) or factors essential for methicillin resistance such as femA (Berger-Bächi et al., 1992;Kizaki et al., 1994; Unal et al., 1992; Vannuffel et al., 1995) or femB (Berger-Bächi et al., 1992; Kizaki et al., 1994). On the other hand, different studies have also shown the applicability of PCR to the detection of staphylococcal antibiotic resistance genes (Anthony et al., 1999; Berger-Bächi et al., 1992). In order to accelerate the procedure of identification in clinical microbiology laboratories, it is very important to havea simple and rapid method for DNA extraction. There are several reports in the literature describing methods of extracting DNA from overnight liquid cultures (Nunes et al., 1999; Tokue et al., 1992; Vannuffel et al., 1995). Ouraim was to develop a PCR for the simultaneous identification of S. aureus strains and detection of mecA gene using a method of extracting DNA directly from a single colony. For the identification of S. aureus, we employed PCR primers targeted to the specific sequence gene. Indeed, no staphylococcal species would yield a PCR fragment using these primers (Martineau et al., 1998; Meshref et al., 2011).
Currently, multiple-antibiotic-resistant S. aureus strains constitute a major health care problem, since they are the etiologic agent of several nosocomial and community-acquired pathological infections. For that reason, accurate and fast detection of resistant isolates constitutes a critical goal of clinical microbiology, and therefore, PCR assays have become an essential tool in laboratory programs. Although previous reports have evidenced the utility of PCR for the accurate detection of the mecA gene (Tokue et al., 1992; Schmitz et al., 1997) and the possibility of simultaneous identification of S. aureus and detection of mecA (Geha et al., 1994; Salisbury et al., 1996; Towner et al., 1998; Vannuffel et al., 1998; Jonas et al., 1999; Tenover et al., 1999). Furthermore, fast DNA extraction methods have also been reported, but normally they are not performed from a single colony and most of them need the use of lytic enzymes, e.g., lysostaphin, and organic solvents, e.g., phenol-chloroform (Anthony et al., 1999; Vannuffel et al., 1995; Vannuffel et al., 1998).Since the convenience of performing all three PCR amplifications in a single tube and from a single colony is obvious, we focused on optimizing the triplex PCR herein described. Thus, we firstly optimized a quick method of extracting DNA from a single colony which yielded enough DNA for optimal PCR amplification without the need of overnight liquid culture, a lytic enzyme, or an organic solvent and without PCR bias due to inhibitory substances. When PCR was performed using DNA obtained by this method or by previously reported ones (Anthony et al., 1999; Jonas et al., 1999; Nunes et al., 1999).
For the strains included in this study, microbiological susceptibility testing and PCR results did not show concordant results (Table 2). This is comparable to Olsson et al. (1993) and Cassettari et al. (2005). Detection of the mecA gene was considered the gold standard for MRSA confirmation (Chambers, 1997; Skov et al., 2006). The mecA gene, which is responsible for this resistance, is often associated in-vitro with resistance to all ß-lactam antibiotics. MRSA strains are frequently resistant to other classes of antibiotics and results compared with conventional methods of MRSA detection. Previous studies have reported discrepancies, noting that some strains lacking mecA displayed phenotypic resistance to methicillin while others containing mecA showed phenotypic susceptibility (Araj et al.,1997; Frebourg et al., 1998; Kohner et al.,1999; Wallet et al., 1996 ). Additionally, mecA transcriptional activity does not correlate with phenotypic methicillin resistance (Niemeyer et al., 1996).For these reasons; we believe that only a microbiological susceptibility testing approaches should not be used as a reliable identification of mecA gene in MRSA isolates.
5. Conclusion
Nowadays, with only a few antibiotics such as vancomycin constituting the last defense against MRSA, and due to the increasing incidence and spread of MRSA, it is absolutely necessary that fast and sensitive laboratory methods are available for the immediate detection of multiple-antibiotic-resistant MRSA. This method reliably amplified DNA from Staphylococcus aureus colonies without a DNA extraction step. As this results showed, the method herein described is highly sensitive, specific, fast, and feasible. Hence, considering that it represents a rapid, simple, and cost-effective method, it could be systematically applied in clinical microbiology laboratories for the identification of MRSA, bringing insights into antibiotic therapy design and helping treatment to be initiated without delay.
Acknowledgments
We are grateful to the management of the eight health institutions that participated in the study for their ethical permission to collect bacterial isolates from their facilities. Our appreciation also goes to the entire staff of medical microbiology laboratories of the various health institutions for their valuable contributions and assistance in the collection of the Staphylococcus aureus isolates. We gratefully thank Inqaba Biotechnical Industries (Pty) Ltd (South Africa) for providing the oligonucleotide primers. The molecular biology works were all carried out in the Molecular Biology Laboratory of the department of Biochemistry, Bayero University Kano. We would like to thank the department of Biochemistry and the management of Bayero University Kano for the establishment of the Molecular Biology Laboratory. We also acknowledge the technical staff of Biochemistry department for their technical assistance.
References
- Alarcón, T., J. C. Sanz, F. Blanco, D. Domingo, and M. López-Brea (1998). High-level mupirocin resistance among Spanish methicillin-resistant Staphylococcus aureus. Eur.J. Clin. Microbiol. Infect. Dis. 17:877-879.
- Amábile-Cuevas, C. F (1993). Molecular Biology Intelligence Unit. Origin, evolution and spread of antibiotic resistance genes. R. G. Landes Company, Austin, Tex.
- Anna-Kaarina, J., SannaLaakso, P.,AnnesAittakorpi, M., Lindfors, L., Huopaniemi, H. and Minna, M.(2009).Rapid identification of bacterial pathogens using a PCR- and microarray-based assay. BMC Microbiology 9:161.
- Anthony, R. M., A. M. Connor, E. G.M. Power, and G. L. French (1999). Use of the polymerase chain reaction for rapid detection of high-level mupirocin resistance in staphylococci. Eur. J. Clin. Microbiol. Infect. Dis. 18: 30-34.
- Bej, A. K., M. H. Mahbubani, R. Miller, J. L. Dicesare, L. Haff, and R. M. Atlas (1990). Multiplex PCR amplification and immobilized capture probes for detection of bacterial pathogens and indicators in water. Mol. Cell. Probes 4: 353-365.
- Berger-Bächi, B., A. Strassle, J. E. Gustafson, and F. H. Kayser (1992). Mapping and characterization of multiple chromosomal factors involved in methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 36:1367-1373.
- Bignardi, G. E., Woodford, N., Chapman, A., Johnson, A. P. and Speller, D. C. E (1996). Detection of the mec-A gene and phenotypic detection of resistance InStaphylococcusaureus isolates with border line or low-level methicillin Resistance. Journal of Antimicrobial Chemotherapy 37: 53—63.
- Brakstad, O. G., K. Aasbakk, and J. A. Maeland (1992). Detection of Staphylococcus aureus by polymerase chain reaction amplification of the nuc gene. J. Clin. Microbiol.30:1654-1660.
- Chambers, H. F. 1997. Methicillin resistance in staphylococci: molecular and biochemical basis and clinical implications. Clin. Microbiol. Rev. 10:781-791.
- Cockerill, F. R (1999). Genetic methods for assessing antimicrobial resistance .Antimicrob. Agents Chemother.43:199-212.
- Dyke, K. G., S. P. Curnock, M. Golding, and W. C. Noble (1991). Cloning of the gene conferring resistance to mupirocin in Staphylococcus aureus. FEMS Microbiol. Lett. 61:195-198.
- Geha, D. J., J. R. Uhl, C. A. Gustaferro, and D. H. Persing (1994). Multiplex PCR for identification of methicillin-resistant staphylococci in the clinical laboratory. J. Clin. Microbiol. 32:1768-1772.
- Hashimoto, H., M. Inouye, and I. Hayashi (1994). A survey of Staphylococcus aureus by typing and drug-resistance in various areas of Japan during 1992 and 1993. Jpn. J. Antibiot. 47:618-626.
- Jonas, D., H. Grundmann, D. Hartung, F. D. Daschner, and K. J. Towner (1999). Evaluation of the mecAfemB duplex polymerase chain reaction for detection of methicillin-resistant Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis.18:643-647.
- Kizaki, M., Y. Kobayashi, and Y. Ikeda (1994). Rapid and sensitive detection of the femA gene in staphylococci by enzymatic detection of polymerase chain reaction (ED-PCR): comparison with standard PCR analysis. J. Hosp. Infect. 28:287-295.
- Kobayashi, N., H. Wu, K. Kojima, K. Taniguchi, S. Urasawa, N. Uehara, Y. Omizu, Y. Kishi, A. Yagihashi, and I. Kurokawa (1994). Detection of mecA, femA and femB genes in clinical strains of staphylococci using polymerase chain reaction. Epidemiol. Infect. 113:259-266.
- Livermore, D. M (2000). Antibiotic resistance in staphylococci. Int. J. Antimicrob. Agents16:3-10.
- Martineauf, P., Roy, P.M. and Bergeron, M.G. (1998). Species-specific and ubiquitous-DNA based assay for rapid identification of staphylococcus aureus. Clin. Microbiol.36: 618-623.
- Martinez, J. L., and F. Baquero (2000). Mutation frequencies and antibiotic resistance. Antimicrob. Agents Chemother. 44:1771-1777.
- Méndez-Álvarez, S., X. Pérez-Hernández, and F. Claverie-Martín (2000). Glycopeptide resistance in enterococci. Int. Microbiol. 3:71-80.
- Murray, P. R., E. J. Baron, M. A. Pfaller, F. C. Tenover, and R. H. Yolken (ed.) (1999). Manual of clinical microbiology, 7th ed. ASM Press, Washington, D.C.
- NCCLS (2001). Performance standards for antimicrobial disk susceptibility tests. Approved standard M2-A7, 7th ed. NCCLS, Wayne, Pa.
- Nunes, E. L., K. R. dos Santos, P. J. Mondino, M. D. Bastos, and M. Giambiagi-de Marval (1999). Detection of ileS-2 gene encoding mupirocin resistance in methicillin-resistant Staphylococcus aureus by multiplex PCR. Diagn. Microbiol. Infect. Dis. 34:77-81.
- Perez, R. E., Claverie, M. F., Villar, J. and Méndez, Á. (2001).Multiplex PCR for Simultaneous Identification of Staphylococcus aureus and Detection of Methicillin and Mupirocin Resistance. Journal of Clinical Microbiology39 (11): 4037-4041.
- Ramsey, M. A., S. F. Bradley, C. A. Kauffman, and T. M. Morton (1996). Identification of chromosomal location of mupA gene, encoding low-level mupirocin resistance in staphylococcal isolates. Antimicrob. Agents Chemother. 40:2820-2823.
- Salisbury, S. M., L. M. Sabatini, and C. A. Spiegel (1996). Identification of methicillin-resistant staphylococci by multiplex polymerase chain reaction assay. Microbiol. Infect. Dis. 107:368-373.
- Schmitz, F. J., C. R. Mackenzie, B. Hofmann, J. Verhoef, M. Finken-Eigen, H. P. Heinz, and K. Kohrer (1997). Specific information concerning taxonomy, pathogenicity and methicillin resistance of staphylococci obtained by multiplex PCR. J. Med. Microbiol.46:773-778.
- Shopsin, B., M. Gomez, M. Waddington, M. Riehman, and B. N. Kreiswirth (2000). Use of coagulase gene (coa) repeat region nucleotide sequences for typing of methicillin-resistant Staphylococcus aureus strains. J. Clin. Microbiol. 38:3453-3456.
- Tang, Y. W., G. W. Procop and D. H. Persing (1997). Molecular diagnostics of infectious diseases.Clin. Chem. 43:2021-2038.
- Tenover, F. C., R. N. Jones, J. M. Swenson, B. Zimmer, S. McAllister, J. H. Jorgensen, and the NCCLS Staphylococcus Working Group (1999). Methods for improved detection of oxacillin resistance in coagulase-negative staphylococci: results of a multicenter study. J. Clin. Microbiol. 37:4051-4058.
- Tokue, Y., S. Shoji, K. Satoh, A. Watanabe, and M. Motomiya (1992). Comparison of a polymerase chain reaction assay and conventional microbiologic method for detection of methicillin-resistant Staphylococcus aureus. Antimicrob.AgentsChemother. 36:6-9.
- Towner, K. J., D. C.S. Tabot, R. Curran, C. A. Webster, and H. Humphreys (1998). Development and evaluation of a PCR-based immunoassay for the rapid detection of methicillin-resistant Staphylococcus aureus. J. Med. Microbiol. 47:1-7.
- Unal, S., J. Hoskins, J. E. Flokowitsch, C. Y. Wu, D. A. Preston, and P. L. Skatrud (1992). Detection of methicillin-resistant staphylococci by using the polymerase chain reaction. J. Clin. Microbiol. 30:1685-1691.
- Vannuffel, P., J. Gigi, H. Ezzedine, B. Vandercam, M. Delmee, G. Wauters, and J. L. Gala.(1995). Specific detection of methicillin-resistant Staphylococcus species by multiplex PCR. J. Clin. Microbiol.33:2864-2867.
- Vannuffel, P., P. F. Laterre, M. Bouyer, J. Gigi, B. Vandercam, M. Reynaert, and J. L. Gala.(1998). Rapid and specific molecular identification of methicillin-resistant Staphylococcus aureus in endotracheal aspirates from mechanically ventilated patients. J. Clin. Microbiol.36:2366-2368.
- Voss, A., D. Milatovic, et al.(1994). Methicillin-resistant Staphylococcus aureus in Europe. Eur. J.Clin. Microbiol. Infect. Dis.13:50-55.



