
International Journal of Bioinformatics and Biomedical Engineering, Vol. 2, No. 5, September 2016 Publish Date: Sep. 10, 2016 Pages: 59-64
Putative Promoters of Two Complete Zika Virus Genome
Md. Zakir Hossain1, *, Rozina Akter2
1Department of Pharmaceutical Sciences, Biomanufacturing Research Institute and Technology Enterprise (BRITE), North Carolina Central University, Durham, North Carolina, USA
2BioMedNano Research Institute, Little Rock, Arkansas, USA
Abstract
The recent outbreak of Zika virus (ZikaV), which is transmitted through Aedes mosquitoes, is an emerging arbovirus, poses global health concern. Currently, there are no effective vaccinations or proven therapeutics that specifically targets the complete ZikaV genomes. To design an effective antiviral therapeutics for ZikaV, the complete genomes should be targeted along with its possible functions. Since promoters are most important regulatory regions for gene expression, identification of putative promoters are vital. The promoter is a short segment of DNA sequence where RNA polymerase first attaches. It forms a recognition and binding site for the RNA polymerase. In addition, it is asymmetrical and thus indicating the site of initiation and direction of transcription. To date, there is no reported data available for the identification and characterization of promoter sequence in complete ZikaV genomes. By considering the limited data on putative promoter motifs of complete ZikaV genome, our study was designed to identify, characterize and investigate the putative promoter motifs in two complete Zika viral strains (ZikaV isolate SSABR1 and second, Brazil-ZKV2015). In fact, the promoter sequences were identified in both of the two complete ZikaV genomes. Further, the significant promoters name, sequence, weight and locations were also noted. Thus, in silico identification of putative promoter motifs in the two complete ZikaV genomes were studied. Therefore, ZikaV promoters study can be helpful to understand the regulation of ZikaV genes and their functions, which eventually will lead to develop live attenuated ZikaV vaccines and gene therapy.
Keywords
ZikaVirus, Genome, Promoters, Transcription Factors, Vaccines, Gene Therapy
Received:April 26, 2016
Accepted: May 9, 2016
Published online: September 10, 2016
@ 2016 The Authors. Published by American Institute of Science. This Open Access article is under the CC BY license. http://creativecommons.org/licenses/by/4.0/
Contents
1. Introduction 2. Methodology 2.1. Retrieval of Two ZikaV Genome Sequences 2.2. Sequence Similarity Search and BLAST Tree View 2.3. Analysis of Two Complete ZikaV Genomes 2.4. The G+ C content 2.5. Putative Promoter Determination Based on Transcription Start Site 3. Results and Discussions 3.1. Analysis of GC Content Zika Virus Isolates SSABR1 and Brazil-ZKV2015 3.2. Prediction of Promoter Sequences in Two Complete ZikaV Sequences 4. Conclusion
1. Introduction
The ZikaV is an enveloped, icosahedral, single-stranded positive (+ ve) sense virus and belongs to the family Flaviviridae, and Genus Flavivirus [1,2]. Though discovered long ago (1947), the circulation of ZikaV in Ocenia had been reported during 2013-2014 [3]. Moreover, ZikaV has been very recently (2015) associated with severe birth defects responsible for abnormally small heads (microcephaly) in new born babies and an auto-immune disorder named Guillan-Barre Syndrome (GBS) [4-6]. Now, Zika virus has been pandemic and is very rapidly circulating in 26 countries across America and throughout the world [7]. By carefully examining and analyzing the complete ZikaV genomes and its promoters, we can identify the transcriptions Factors (TFs) of ZikaV that are primarily responsible for ZikaV associated diseases. The promoter is a regulatory region of DNA located upstream towards the 5' region of the sense strand that initiates transcription of a particular gene. They are located near the transcription start sites (TSS) of genes, on the same strand and can be about 100–1000 base pairs long [8]. The core promoter includes the TSS and elements directly upstream. The proximal promoters are the sequences upstream of the genes that tend to enclose regulatory elements. The internal promoter are a class of enhancers that are gene-specific sequences and increase transcription. The strong promoters match consensus sequence closely i.e. operons transcribed efficiently and on the other hand the weak promoters match consensus sequence poorly i.e. operons transcribed infrequently [9].
Identification of promoters and transcription factor (TF) sites are necessary to understand the function and regulation of genes. The promoter is a most important regulatory region that controls and regulates gene expression at the transcription level. The promoter is necessary to start transcription [8]. The viral promoters are generally used as regulatory elements in gene therapy vectors because of its strong activities in various cell lines in vitro. The promoters control the binding of the RNA polymerase and transcription factors (TFs). Thus, it has a huge role in determining where and when the gene of interest can be expressed [10]. Presently, there is no proven therapeutics or effective vaccines available against the ZikaV. In this study, we have identified, analyzed and provided a brief description and functions of putative promoters of two complete ZikaV genomes
2. Methodology
2.1. Retrieval of Two ZikaV Genome Sequences
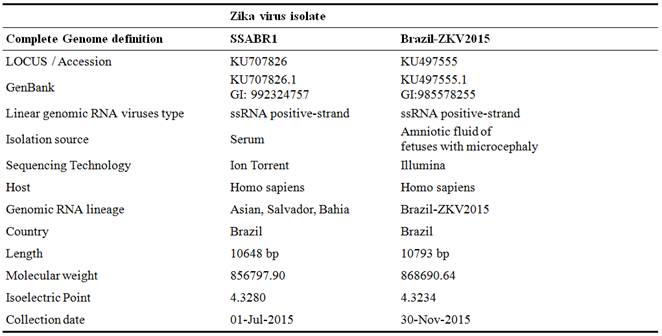
The complete genome sequences of two ZikaV were selected. First, ZikaV isolate SSABR1 (GenBank Accession KU707826.1) and the second, Brazil-ZKV2015 (GenBank Accession KU497555.1). The two complete ZikaV nucleotide sequences were obtained from the biological databases of National Centre for Biotechnology Information which is cited at http://www.ncbi.nlm.nih.gov/genomes/VIRUSES/viruses.html [11].
2.2. Sequence Similarity Search and BLAST Tree View
The relatedness of the selected two ZikaV sequences were evaluated by BLAST (Basic Local Alignment Search Tool) implemented via the NCBI website (www.ncbi.nlm.nih.gov/blast) [12].
2.3. Analysis of Two Complete ZikaV Genomes
The genome-size of the two ZikaV species were analyzed in FASTA format extracted from NCBI Genbank database.
2.4. The G+ C content
The GC content was compared and the two complete genomes were analyzed. (http://www.sciencebuddies.org/science-fair-projects/project_ideas/Genom_GC_Calculator.shtml [13].
2.5. Putative Promoter Determination Based on Transcription Start Site
The putative promoters in the genome of two ZikaV species were identified using the PROMOTERSCAN (PSCan) program at http:// www.bimas.cit.nih.gov/molbio/proscan/. The analysis is done using the PSCan Version 1.7 suite of programs [9].
Figure 1. Schematic representation of prediction of putative promoter on complete Zika viral genome.
3. Results and Discussions
The analyzed two complete Zika Virus genomes used in this research are depicted in Table 1.
Table 1. Analysis of Zika virus isolates SSABR1 and Brazil-ZKV2015.
NCBI Blast tree view was generated by using BLAST pairwise alignments and found that the selected two complete ZikaV genome strains are closely related.
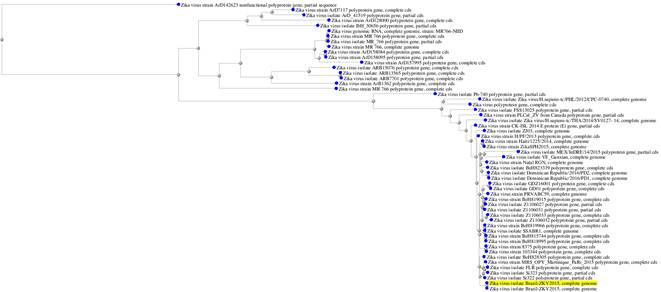
Figure 2. Zika virus isolate Blast tree view.
3.1. Analysis of GC Content Zika Virus Isolates SSABR1 and Brazil-ZKV2015
The GC base pairs are more stable than AU base pairs, due to having three hydrogen bonds and AU on the contrary have two. This makes high GC-content RNA structures more tolerant to high temperatures. The genomic comparisons of G-C % of the two ZikaV are almost same 51.2% and 51.3% (Figure 3 and Table 2).
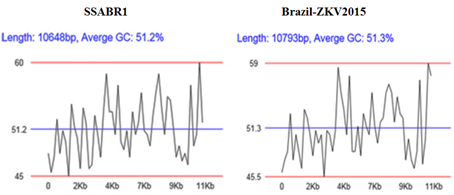
Figure 3. Graphical analysis of GC content.
Table 2. Analysis of GC content Zika virus isolates SSABR1 and Brazil-ZKV2015.
3.2. Prediction of Promoter Sequences in Two Complete ZikaV Sequences
Using the PScan version 1.7, the significant promoters signal of two complete ZikaV genome of SSABR1 and Brazil-ZKV2015 were predicted. The PScan program was used to identify the putative promoter in the complete genome of two ZikaV sequences. The PScan program generally includes three databases viz TF database, promoter database and non-promoter set. The PScan finds putative ZikaV promoter sequences in primary sequence data. The predicted promoter sequences are the regions of ZikaV DNA that contains a significant number and type of transcriptional elements (TEs) that are usually associated with Pol II promoter sequences. The PScan was set a predetermined cutoff score to identify 70% of primate promoter sequences in the eukaryotic promoter database. In general, at this cutoff score, the false positive predictions normally occur at a rate of approximately one in every 14,000 single strand bases [9]. The program reported, both the TATA box position and the location of estimated TSS is within + / - 10 bases of the actual TSS. Significant signals (most of them transcriptional elements) are also noted.
The PScan program identified promoters from Zika viral isolate SSABR1 complete genome. The Zika viral isolate SSABR1 has total processed sequence of 10648 base pairs. The promoter region predicted on forward strand in 7955 to 8205. The promoter score was found 58.72 where the promoter cutoff value was 53.000000. The TATA was found at 8177, and the estimated TSS was 8207. On the other hand, the Brazil-ZKV2015 processed sequence were 10793 base pairs. The promoter region predicted on forward strand 7977 to 8227 and the promoter score was 55.23. The TATA found at 8199, whereas estimated TSS 8229 and the promoter cutoff value was 53.000000.
The Figures 4 and 5 exhibiting the location of predicted promoter sequences in two complete ZikaV sequences. The bright green lines indicate positive strand and the red lines indicate negative strand. The yellow-orange bright box within positive strand and blue box within negative strand indicates promoter names together with their respective sequences. The number below the box indicates the specific location of the promoter. The letters above the box indicates the promoter designation. The number above the box (within bracket) indicates their corresponding weight.
Table 3. The SSABR1significant signals.
Figure 4. Prediction of putative promoter on Zika viral complete genome, SSABR1.
Table 4. The Brazil-ZKV2015 significant signals.
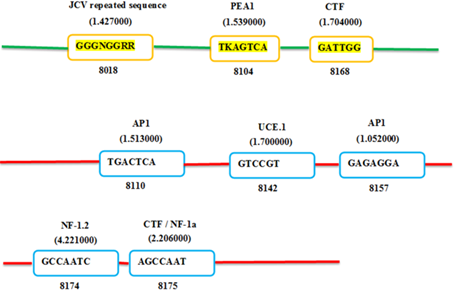
Figure 5. Prediction of putative promoter on Zika viral genome of BRAZIL-ZKV2015.
The identified putative promoters of two complete ZikaV genomes are discussed with their associative functions. The John Cunningham Virus (JCV repeated sequence) is a ubiquitous human pathogen known as human polymavirus or papovavirus causes fatal progressive multifocal leuko-encephalopathy (PML) in the brain. The JCV repeated sequence has been found in human neurotropic papovirus within 98 bp specific transcription of early and late stages of viral promoter sites [14]. The A enhancer of the polyomavirus (PEA-1) is a key components of the polyomavirus late transcription initiator element. It is 110-bp domain. On late transcription only PEA1 acts positively and inactivation of the NF-D site is without any effect [15-16]. Activating Protein-1(AP-1) is a transcription factor collectively known as AP1 is composed of Jun, Fos or ATF subunit. Ap1 binds to a common sequence specific DNA site. To perform unique biological function, different AP1 factors may regulate different target genes [17]. The upstream control element (UCE) is a key element which extends 100 to 150 upstream of the TSS. It is playing an important role in rRNA transcription and is recognized by RNA polymerase 1 [18]. The CTF stands for CCAAT Transcription Factor and the CTF/NF-1 stands for CAAT-binding Transcription Factor / Nuclear Factor-1(NF-1). It is composed of polypeptides encoded by four paralogous genes located on different chromosomes in mammals. Such as NF1A, NF1B, NF1C and NF1X. It binds as dimer, and preferred binding palindrome sequence. Nuclear Factor-1 (NF-1) which is also known as NF-1 / CTF, is ubiquitously found in most tissues and contain a constitutive DNA binding gene harbors a hypo-methylated CpG island. It is responsible for the development of different types of neuro-fibromas and malignant transformation through methylation [19].
In molecular biology, retroviral TATA box is also called as Goldberg-Hogness box. It is (-) 10 bp upstream from transcription start site (TSS), and is rich in A-T bp. Due to weaker base-pairing interactions between A and T bases, as compared to G-C, AT-rich sequence of the TATA box facilitates easy unwinding [8]. In our study, based on TATA box, the Promoter scan program estimates the TSS position from the TATA box location. Transcription Factor II D (TFIID) is one of the common transcriptions factor or promoter site that is prerequisite for the RNA polymerase II pre-initiation complex. TFIID consists of TATA-binding proteins (TBP) and TBP associated Factors (TAFs) that play significant role in both positive and negative transcription regulation. TFIID binds to TATA box in the core promoter of a particular gene. It then regulates the activities of more than 70 polypeptides required for the transcription initiation by RNA polymerase [20]. Thus, it also acts as channel for regulatory signals [21].
The significant signals of SSABR1 and Brazil-ZKV2015 have common promoters though their locations are different. The complete genome of SSABR1 has two extra promoters named retroviral TATA and TFIID.
4. Conclusion
To conclude, in the present study, for the first time, we have successfully identified ZikaV promoter sites in two ZikaV complete genomes using bioinformatics tools. Thus, the purpose of this study was to identify and analyze the putative promoter motifs in the two complete ZikaV genome. Identification of ZikaV promoters are beneficial in designing of ZikaV expression vectors. This study can be used to understand the regulation of ZikaV gene expression. Finally, the prediction of putative promoters in complete ZikaV genome is particularly desirable to have a major impact upon ZikaV drugs discovery as well as possible gene therapy through site directed mutagenesis. Ultimately, such study has enormous implications in ZikaV infections and leads to develop major ZikaV vaccines.
References
- Kruger, R. P. (2016). Zika Virus on the move. Cell Leading Edge Select, 164, February 11, 2016.
- Faye, O., Freire, C.C., Iamarino, A., Faye, O., de Oliveira, J.V., Diallo, M., et al.(2014). Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis, 8 (2014), p. e263.
- Bogoch, I.I., Brady, O.J., Kraemer, M.U., German, M., Creatore, M.I., Kulkarni, M.A., Brownstein, J.S., Mekaru, S.R., Hay, S.I., Groot, E., Watts, A, Khan, K.(2016) Anticipating the international spread of Zika virus from Brazil. Lancet Jan 14) pii: S0140-6736(16)00080-5.
- Campos, G.S. Bandeira, A.C. Sardi, S.I. (2015). Zika virus outbreak, Bahia, BrazilEmerg Infect Dis, 21 (2015), pp. 1885–1886.
- Oliveira M.A.S., Malinger, G., Ximenes, R.,Szejnfeld, P.O., Sampaio, S. A., Filippis, M.B., (2016). Zika virus intrauterine infection causes fetal brain abnormality and microcephaly: tip of the iceberg? Ultrasound in Obstetrics & Gynecology, 47, pp. 6–7.
- Oehler, E., Watrin, L., Larre, P., Leparc-Goffart, I., Lastere, S., Valour, F., et al. (2014). Zika virus infection complicated by Guillain-Barre syndrome--case report, French Polynesia, December 2013, Euro Surveill, 19 (2014), p. 20720.
- Petersen E., Wilson M.E., Touch S, McCloskey, B., Mwabae, P. Batese M, Dard, O., Mattes, F., Kidd, M., Ippolitog, G., I. Azharh, E.I., Zumla, A. (2016). Rapid Spread of Zika Virus in the Americas - Implications for Public Health Preparedness for Mass Gatherings at the 2016 Brazil Olympic Games, International Journal of Infectious Diseases, Volume 44, Pages 11–15.
- Lewin B, Genes VIII. 2004 pp. 636–637. ISBN 0-13-144946-X.
- Prestridge, D.S. (1995). Predicting Pol II Promoter Sequences Using Transcription Factor Binding Sites. J. Mol. Biol. 249: 923-32.
- Somvanshi, P., Singh, V., and Seth, P.K., (2008). Identification and analysis of putative promoter motifs in Flavivirus genome, Bioinformation. 3(4): 162–167.
- "National Centre for Biotechnology Information". http://www.ncbi.nlm.nih.gov/nuccore/992324757, May 03, 2016.
- "National Centre for Biotechnology Information." http://www.ncbi.nlm.nih.gov/nuccore/KU497555, May 04, 2016.
- "Sciencebuddies". http://www.sciencebuddies.org/science-fair-projects/project_ideas/Genom_GC_Calculator.shtml, May 02, 2016.
- Hiroomi T. (1991). Regulation of JCV promoter function: Evidence that a penta-nucleotide "silencer" repeat sequence AGGGAAGGGA down-regulates transcription of the JC virus late promoter. Virology. 1991; 180: 327.
- Shivakumar CV, Das, GC. (1998) The A enhancer of polyomavirus: protein-protein interactions for the differential early and late promoter function under nonreplicating conditions. Intervirology. 1998; 41(2-3):103-9.
- Yoo W, Martin ME, Folk WR. (1991). PEA1 and PEA3 enhancer elements are primary components of the polyomavirus late transcription initiator element.J Virol. 1991 Oct; 65(10):5391-400.
- Hess J, Angel P, Schorpp-Kistner M. 2004AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. Dec 1; 117(Pt 25): 5965-73.
- Ghoshal, K., Majumder, S., Jharna, D., Motiwala, T., Bai, S., Sudarshana M. S., Frankel, W., and Jacob. S. T., (2004). Role of Human Ribosomal RNA (rRNA) Promoter Methylation and of Methyl-CpG-binding Protein MBD2 in the Suppression of rRNA Gene Expression. J Biol Chem. Feb 20; 279(8): 6783–6793.
- Horan MP, Cooper DN, Upadhyaya M. Hypermethylation of the neurofibromatosis type 1 (NF1) gene promoter is not a common event in the inactivation of the NF1 gene in NF1-specific tumours. Hum Genet. 2000 Jul; 107(1): 33-9.
- Kurjogi MM, Sanakal RD, Kaliwal BB. Identification and analysis of putative promoter motifs in bovine herpes virus. Bioinformation. 2012; 8(23): 116770.
- Hale TK, Braithwaite AW. The adenovirus oncoprotein E1a stimulates binding of transcription factor ETF to transcriptionally activate the p53 gene. J Biol Chem. 1999 Aug 20; 274(34):23777-86.



